Jerry Bergman
Z časopisu Creation Research Society Quarterly, roč. 41, č. 4, březen 2005, str. 265 – 272 přeložil M. T. – 12/2007
Ve zkratce
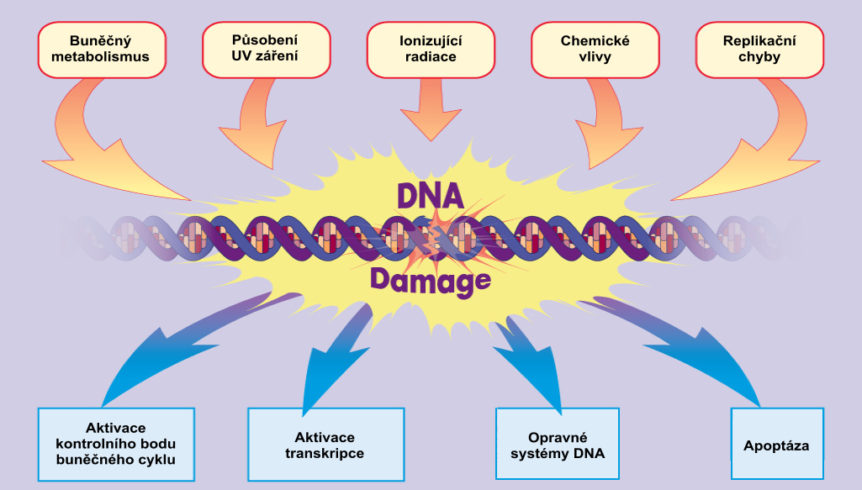
Nejdůležitějším zdrojem vší rozmanitosti, s nímž může pracovat přírodní výběr, jsou genetické mutace. Existují četné buněčné opravné systémy, jež zajišťují, aby exprese mutace byla neobyčejně vzácná. Tyto opravné systémy představují velké problémy pro evoluci, protože prakticky všechny genetické inovace způsobené změnami párů bází budou opraveny (a tedy se neprojeví) či bude buňka jako taková zničena. Kdyby byly genetické opravné systémy dokonalé, pak je jasné, že by veškerá makroevoluce byla nemožná. Tyto genetické opravné systémy mluví proti makroevoluci, alespoň proti té, která je vypůsobena nahromaděním mutací.
Úvod
Evoluční naturalizmus učí, že stvořitelem všech forem života jsou koneckonců jen chyby v replikaci DNA známé jako genetické mutace. Jak tvrdí Eldredge (1982):
„jedinou skutečně významnou silou stojící za všemi genetickými (a tedy evolučními) změnami je přírodní výběr… genetická změna je do značné míry funkcí přírodního výběru pracující na poli různorodosti, jak ji nabízí každá generace. Nové rysy se čas od času objeví následkem mutací. Většina mutací je zhoubná; některé jsou neutrální, či dokonce přínosné. Neutrální či přínosné mutace přetrvávají, a jednoho dne se mohou ukázat jako skutečná výhoda, když okolí začne klást na organizmy nové nároky… A je-li dostatek času – nezapomeňte, že geologové nám říkají, že země je stará celých 4,5 miliardy let – , dojde ke všem typům změn.“ (str. 69).
Problém evoluce cestou mutací byl shrnut takto:
„Evoluci mutačních rychlostí řídí dvě protikladné síly: náklady na zhoubné mutace a schopnost přizpůsobit se měnícímu se prostředí. Na rozdíl od asexuálních populací, výběr působící v populaci sexuální vždy upřednostní mutační poměr nula“. (McVean a Hurst, 1997, str. 388).
Zhoubné mutace mohou způsobovat nejen značné zdravotní problémy, ale také mohou vést k vymření druhu, pokud nejsou opraveny. „Pokud nejsou tyto chyby zachyceny v počátcích, mohou se škodlivé mutace hromadit, takže bílkoviny k opravám chyb a nevhodných spojení jsou životně důležité“ (Kolodner, 2000, str. 678). Počet mutací je podstatně omezován jak vysokou spolehlivostí při replikaci DNA, tak četnými vysoce efektivními opravnými systémy. V závislosti na specifickém organizmu, jeho prostředí, a jiných faktorech jsou poměry neopravených mutací odhadovány na pouhých asi 10–5 na gen a jedno buněčné dělení (Puck et al., 1996;1998; Mader, 1998), i když takové odhady budou kolísat v závislosti na použité metodě.
Mader (1998) konstatoval, že proces korektur je tvořen mechanizmem, který dosahuje tak vysoké míry přesnosti, že „nakonec dojde jen k jedné chybě na každou miliardu replikovaných nukleotidových párů“ (str. 247). Jiné odhady se liší, ale panuje všeobecná shoda, že poměr je extrémně nízký. Přesnost korektur v tomto systému omezuje poměr chyb v reprodukci odhadem od asi jedné chyby na 100 000 párů bází až k asi jedné ku 10 miliardám párů bází – podivuhodně vysoká míra přesnosti (Radman a Wagner, 1988).
Tato nízká úroveň chyb je dokonce ještě dále omezena několika opravnými mechanizmy vestavěnými do genetického systému všech jedno- i mnohobuněčných organizmů a zajišťujícími, že chyby jsou velmi vzácné. Dokonce i organizmy jako viry (jež nemají opravné enzymatické systémy) jsou do jisté míry chráněny, protože využívají buněčné enzymy svého hostitele pro opravu mutací. Arzenál opravných mechanizmů je tak přesný, že opraví odhadovaných 99,9 procenta původních chyb (Friedberg, 2003; Jorde et al., 1997; Yang et al., 1996; Radman a Wagner, 1988). A ty mutace, jež nejsou opraveny, jsou často fixovány udržovacími mechanizmy buňky (Ridley, 2001; Harwood a Meuth, 1995).
Výsledkem fungování těchto opravných systémů je fakt, že věrnost replikace je tak vysoká, že se objevují méně než odhadované tři chyby během replikace celé lidské bytosti (průměrně tři miliardy párů bází). Tuto úroveň „fenomenální přesnosti zajišťuje několik DNA reparačních enzymů včetně některých forem DNA polymerázy, které „korigují“ každý dceřinný řetězec během syntézy i po ní“ (Audesirk a Audesirk, 2000, str. 130).
DNA polymeráza = enzym, který katalyzuje tvorbu DNA z prekurzorů, za přítomnosti již existující DNA jako šablony (pozn. překl.).
Směrovost DNA umožňuje korekturám i opravným enzymům rozpoznat rodičovský řetězec, a pak identifikovat nevhodná spojení a nakonec opravit řetězec dceřinný, který běží v opačném směru. Tento opravný systém je nutný, protože „aby mohl organizmus fungovat, musí bránit integritu svého genetického materiálu. Důvod je prostý: hromadění poškozené DNA může vést ke škodě včetně rakoviny“ (Marx 1994b, str. 1321).
Přesnost oprav DNA naznačuje studie u 76 000 potomků lidí přeživších výbuchy atomových bomb v Hirošimě a Nagasaki, kteří byli vystaveni masivnímu množství radiace. Studie shledala, že „žádný změřitelný efekt nemohl být zjištěn u zárodečných buněk a opravy DNA jsou za to alespoň zčásti odpovědné“ (Jorde et al., 1997, str. 37). Bylo tomu tak, přestože byly u somatických buněk přeživších lidí nalezeny závažné důkazy o účincích záření.
Tento fakt je rozhodující ve sporu o počátky života, protože neodarwinizmus vyžaduje mutační změny po zárodečné linii buněk, a studie ukazují, že gamety jsou obzvlášť odolné vůči mutacím měnícím páry bází.
Předcházení mutacím: chráněný kód
O genetickém kódu se říká, že je chráněný – znamená to, že třetí písmeno většiny kódů aminokyselin se může změnit bez účinku na samotný kód. Příkladem je GGU, GGC, GGA a GGG, což jsou všechno kódy pro glycin. V souladu s tím, pokud mutace změní třetí bázi v jakémkoli případě, glycin stejně bude kódován – což vylučuje či minimalizuje účinek mnoha bodových mutací (Ritter, 1996).
Z tohoto důvodu mutace ve třetí bázi kodonu často nemění to, která aminokyselina bude umístěna do syntetizovaného polypeptidu. Dále, dojde-li k mutaci ve třetí pozici, výsledkem je často aminokyselina s podobnou funkcí, což umožňuje, aby nová bílkovina měla totéž (či velmi podobné) složení jako předtím. Výsledkem je, že „změna ve třetí bázi kodonu…obvykle vede k novému kodonu pro tutéž aminokyselinu či aminokyselinu podobnou“ (Ritter, 1996, str. 660). Samozřejmě může občas mutace vést k aminokyselině z odlišné rodiny či dokonce ke kodonu terminačnímu. To může vyústit v onemocnění či ve smrt.
kodon = trojice nukleotidů (v mRNA) určující v genetickém kódu jednu určitou aminokyselinu, popř. zahájení či ukončení translace. (pozn. překl.)
Tento mechanizmus (také zvaný kolísavý efekt) umožňuje antikodonům na tRNA napojit se vodíkovým můstkem na více než jeden kodon. V situacích, kdy tRNA se může párovat s několika kodony, všechny tyto kodony specifikují tutéž aminokyselinu. U lidí omezuje kolísavý efekt na 32 počet jádrových tRNA nutných pro přečtení 61 transkribovaných kodonů, což ušetří energii, jež by byla nutná pro shromáždění plné sady 61 tRNA. Tento systém také omezuje nutnou velikost genomu, protože různé geny musí běžně zakódovat každou specifickou tRNA. Také to omezuje dobu nutnou pro sladění kodonu s příslušným antikodonem během translace. Mitochondriální translační mechanizmus umožňuje, že genetický kód v mitochondrii mRNA je dekodován pouhými 24 tRNA.
tRNA = druh ribonukleové kyseliny (angl. transfer RNA), který přenáší aminokyseliny při tvorbě bílkovin v buňce. (pozn. překl.)
mRNA je druh ribonukleové kyseliny, která vzniká přepisem z DNA (transkripcí) a „sestřihem“ (splicing). Z jádra je transportována do cytoplasmy, kde se ve spojení s ribozómy účastní syntézy bílkovin (translace). Přenáší dědičnou informaci, která je uložena v genu a kóduje přesné pořadí aminokyselin v bílkovině (angl. messenger RNA). (pozn. překl.)
Dále, dokonce i změna v první bázi v kódu (pět primárních bází) často – ale jistěže ne vždy – vygeneruje „nový kodon, který kóduje aminokyselinu, jejíž postranní řetězec je podobný v polaritě postrannímu řetězci aminokyseliny specifikované kodonem původním. V důsledku toho polypeptid produkovaný informací v genu s bodovou mutací je buď obecně identický s peptidem normálním či je podobný ve struktuře a biologické aktivitě“ (Ritter, 1996, str. 660). V důsledku toho Ritter (1996) konstatuje, že „polypeptid produkovaný informací v genu s bodovou mutací je obecně identický s peptidem normálním či je podobný ve struktuře a biologické aktivitě. Jelikož většina organizmů nahromadí časem mutace, má ochrana genetického kódu zásadní význam z hlediska přežití“ (str. 660).
Mechanizmus ochrany kódu je také velkým problémem pro makroevoluci, protože zajišťuje, že většina z relativně vzácných neopravených mutací nevyprodukuje aminokyselinu či strukturální změny v bílkovině. Makroevoluce cestou mutací však změny ve tvaru proteinu vyžaduje. Jakmile je jednou přečten iniciační kodon, každá další sekvence nukleotidů obvykle představuje další nedotčený kodon – a je neobvyklé, aby se čtecí rámec posunul, jakmile translace začala (což zaručuje extrémně vysokou věrnost translace). (Ritter, 1996).
U placentálních savců také mnoho gametových mutací nezpůsobí dědičné změny, protože zabrání plodu ve vývoji k narození. Částečně proto tedy asi třetina všech lidských embryí je spontánně potracena, což zajistí, aby většina neopravených projevených mutací v zárodečné linii nebyla předána dále. Mutace, které nejsou neutrální, většinou způsobí nemoc somatické buňky a smrt. Stejně tak mutace u jiných forem života často způsobí, že organizmus zemře v raném stadiu vývoje a tak se zabrání tomu, aby se mutace přenesla do potomstva.
Proofreading (korekce) DNA a reparační systém
Třemi hlavními mechanizmy k omezení chyb jsou vyhýbání se chybám (především díky efektivitě výběru nukleotidů), opravování chyb během replikace DNA (proofreading), a pokročilé opravování chyb k nápravě chyb propuštěných prvními dvěma systémy (Radman a Wagner, 1988).
Replikázy hrají prvořadou roli ve vyhýbání se omylům tím, že přesně řídí párování nukleotidů. Také se účastní čtení korektur, které odstraňuje špatně spárované nukleotidy. Užití primerů (startérů) RNA a jejich automatické odstraňování a nahrazování je dalším mechanizmem k vyhnutí se chybám. Četnost chyb je nepatrně „větší během napojování prvních několika zbytků aminokyselin do nového polypeptidového řetězce“ (Ritter, 1996, str. 725) než během prodlužování řetězce. Výsledkem je, že většina nehlášených chyb je odstraněna během posledních kroků syntézy bílkoviny, kdy se obvykle sestřihuje či modifikuje prvních několik nukleotidů.
Replikáza = RNA-syntetáza, polymeráza, která podporuje syntézu určité RNA v přítomnosti RNA šablony.
Primer = malá molekula potřebná k zahájení syntézy makromolekuly. (pozn. překl.)
Za vysokou věrnost replikace DNA vděčíme také zčásti různým komplexním enzymatickým opravným procesům prováděným samotným komplexem DNA – polymerázy. První úroveň oprav polymerázy je vestavěna do struktury samotného páru bazí DNA. Kód DNA sestává ze čtyř chemických bazí: thyminu, adeninu, cytosinu a guaninu (RNA užívá uracil místo thyminu). Adenin se normálně stabilně váže pouze na thymin či uracil, zatímco cytosin se váže jen na guanin.
Pokud je nukleotidtrifosfát vybraný polymerázou DNA z buněčného zásobníku nukleotidů komplementární s nukleotidem templátu (A je komplementární k T a C ke G), změní se na nukleotidmonofosfát. Energie uvolněná touto přeměnou je použita na svázání oněch dvou nukleotidů vodíkovým můstkem podle templátu poskytnuté nukleotidovým vzorcem. Pokud je navázán nesprávný nukleotid, vazba je nestabilní a nukleotid je většinou vrácen do své trifosfátové formy a uvolněn. Mader (1993) odhaduje, že nesprávně zapojený nukleotid „proklouzne tímto procesem výběru pouze jednou za 100 000 párů bazí“ (str. 243).
Další příčinou tak udivující přesnosti replikace je existence několika rozmanitých opravných systémů, jež využívají několik tuctů různých složitých enzymů (Sutherland a Woodland, 1990). Jak poznamenává Ritter (1996): „Přežití individuálních organizmů i celých druhů závisí na přesné replikaci genetické informace“ (str. 725). Existují různé cesty k opravě DNA, přičemž každá z nich je specializovaná na určitý druh poškození (Culotta a Koshland, 1994). Dále vyrábí většina organizmů četné enzymy, jež mohou opravit jisté druhy poškození DNA. U lidí je známo kolem 50 enzymů, jež se účastní oprav DNA.
Vnitřní chyby, k nimž občas dojde během replikace DNA, jsou obvykle opraveny proto, že komplex polymerázy DNA funguje také jako korektor. Oprava nevhodných zapojení vyžaduje multiproteinový komplex, který rozpozná špatně zapojené báze a použije mnohostranné enzymy k nahrazení nesprávně zapojeného zbytku nukleotidu zbytkem správným. Jedním z faktorů, které mohou spustit opravu, je fakt, že nesprávné zapojení vyprodukuje komplex páru bází o špatném průměru – výsledkem pak je, že špatně zapojený nukleotid způsobí přestávku v replikaci. To spustí proces, který obvykle vyústí ve vyříznutí špatně zapojeného nukleotidu z dceřinného řetězce.
Správným párem je jeden pyrimidin (monocyklická molekula) a jeden purin (bicyklická molekula); nesprávný pár by zahrnoval dva puriny (což vede k páru většímu než je normální) či dva pyrimidiny (což vede k páru tenčímu než je normální). Když se objevila tato úroveň čtení korektur, „poměr chyb je pouze jedna chyba na 10 miliónů párů bazí (Mader, 1993, str. 243). Normálně pouze když se purin spáruje se špatným pyrimidinem (A-C, G-T), může chyba proklouznout, ale i v tomto případě je chyba často opravena jinými opravnými mechanizmy.
Purinové báze = bazické deriváty purinu; 6-aminopurin (adenin) a 2-amino-6-hydroxypurin (guanin). Vyskytují se jako složky nukleotidů a nukleosidů, v nukleových kyselinách slouží k uchování genetické informace.
Pyrimidinové báze = deriváty pyrimidinu sloužící jako stavební součásti nukleových kyselin, zejm. cytosin, thymin, uracil. (pozn. překl.)
Mutace mohou obvykle způsobit trvalé změny párů bazí jen v tom nepravděpodobném případě, že obě báze jsou změněny současně, jako je tomu u dvouřetězcového zlomu. Opravný systém polymerázy DNA užívá původní řetězec DNA, aby zjistil, která báze je správná (kopírovací postup může zjišťovat, která báze je původní a která je dceřinná), a také to, které opravné báze jsou třeba (Ridley, 2001).
Navíc může opravný systém rozlišovat mezi dceřinným (kopírovaným) a rodičovským (původním) řetězcem pomocí mechanizmů jako je jejich rozsah methylace – nově syntetizovaný řetězec DNA není ukončen methylovými skupinami, dokud není zkontrolován (Radman a Wagner, 1988). Rozlišování řetězců dceřinných od rodičovských je důležité, protože nahrazení nukleotidu v řetězci rodičovském by způsobilo rozšíření chyb místo jejich opravy. V důsledku toho, pokud jsou poškozeny oba řetězce molekuly DNA v téže oblasti, je oprava méně pravděpodobná – a pokud se buňka rozdělí a reprodukuje, mutace bude předána dál. Takže k tomu, aby mutace proklouzla tímto opravným mechanizmem, musí se poškození objevit na obou řetězcích DNA – extrémně nepravděpodobná situace.
Methylace DNA = jeden z mechanizmů epigenetické inaktivace genů. Methylován je cytosin. Methylovaná DNA může být reaktivována demethylací. Uplatňuje se v rané embryogenezi, kdy je většina genů vypnuta, tj. nejsou přepisovány. (pozn. překl.)
Dokonce i nejsmrtelnější typ poškození, dvouřetězcové zlomy – kde jsou postiženy obě fosfátové páteře – může být opraven zase jiným opravným mechanizmem (Bartek a Lukas, 2003; Bakkenist a Kasten, 2003). Tento systém zavádí mezimolekulární autofosforylaci několika cílových bílkovin, jež jsou spojeny se signálními sítěmi. Tyto sítě zpomalí postup buňky buněčným dělením a také stimulují opravu dvouřetězcového zlomu (Bartek a Lukas, 2003). Ještě jiný mechanizmus umí opravit chromozom Y. Jelikož tento chromozom nemá svůj protějšek (tj. část chromozomového páru) jako jiný chromozom, má náhradní kopie příslušných genů (Rozen et al., 2003; Skaletsky et al., 2003; Willard, 2003). Chromozom pak vytvoří smyčku, takže defektní gen může použít kopii coby templát k provedení oprav.
Je-li poškozen gen kontrolující přepis DNA a reparační protein není schopen detekovat chyby (čili najít je a opravit), nahromadí se rychle chyby v genech, jež měl kontrolní systém kontrolovat. Pokud jsou nepříznivě postiženy geny, jež brání buňkám nekontrolovaně se dělit, způsobí to často rakovinu. Rakovina tlustého střeva vděčí za svou převahu v typech nádorů tomu, že buňky vystýlající střevo se dělí tak rychle, že celá vystýlka intestinálního traktu je nahrazena asi za tři dny. To nám názorně ilustruje klíčovou roli, kterou sehrává systém kontrolující přepis DNA.
Poškození opravných systémů způsobuje nemoci
Zdá se, že mnoho dalších nemocí vyplývá z defektů v opravném systému, jež umožňují, aby mutace zůstaly neopraveny. Opravný mechanizmus je velmi důležitý pro život, protože škoda způsobená mutacemi může být enormní, a zahrnuje četné nemoci jako třeba rakovinu (Bakkenist a Kasten, 2003; Aldhous, 1995). Například defektní bílkovina k „opravám nesprávných spojení“ způsobuje nepolypózní rakovinu tlustého střeva (Modrich, 1994).
Dědičný defekt zaviněný jedním ze dvou genů zvaných hMLH1 a hMSH 2 (nacházejících se na chromozomech 3 resp. 2) patrně způsobuje až 22 000 případů rakoviny tlustého střeva ročně (Culotta a Koshland, 1993). Tyto dva geny tvoří část reparační dráhy DNA, „jež asi zajišťuje jednu ze základních cest k rakovině, pokud je narušena“, a produkty těchto dvou genů „odstraňují chybná zapojení a instrumentují enzymy, jež působí opravy“ (Service, 1994, str. 1559). Mnoho nemocí jako třeba Fanconiho anemie, ataxie-telangiektazie a Cockayneův syndrom se jeví jako důsledek špatného opravného mechanizmu.
Kontrolní systémy potlačující nádorové bujení
Některé systémy mohou zablokovat reprodukci buněk s poškozenou DNA a řídit jejich opravu, nebo, pokud škoda nemůže být napravena, zničit buňku apoptózou (o níž je řeč dále). Tyhle systémy umějí opravit poškození, jež může způsobit nádory čili rakovinu, a proto jsou zvány tumor-supresorové systémy (systémy potlačující nádorové bujení) . Bílkovina známá jako pRB kontroluje buněčný cyklus, specificky působí jako měnič či přenašeč signálů, které spojují „časovač buněčného cyklu“ s transkripčním mechanizmem (Weinberg, 1995, str. 323). Je-li funkce pRB narušena, je ztracena plná kontrola buněčného cyklu a v důsledku toho je tento důležitý mechanizmus pro kontrolu buněčného dělení také narušen. Z tohoto důvodu slouží pRB k potlačování nádorů, specificky u retinoblastomu a jistých dalších nádorů.
Tumor-supresorový gen = gen, jehož produkt chrání buňky před maligní transformací (antionkogen). Mechanizmus působení těchto genů je různý – podílejí se na dohledu na genom a na jeho opravě, řízení buněčného cyklu aj. Popsána byla již řada těchto genů, k nejznámějším patří p53 a Rb. (pozn. překl.)
Opravný mechanizmus p53
Mezi četnými mechanizmy účastnícími se oprav či prevence škod ze změny fenotypu či z dědičných defektů je nejznámějším příkladem tumor-supresorový gen p53. P 53 hraje životně důležitou roli v regulaci buněčného cyklu v dobách genomického stresu (Wang et al., 1995; Culotta, 1993). Buněčný cyklus sestává ze čtyř hlavních etap. V první, zvané Gap 1 (G-1), dochází k syntéze RNA a bílkovin – jde o přípravu na etapy další. Syntéza DNA probíhá během etapy druhé, zvané Syntéza. Během další fáze, Gap 2 (G-2), se buňka připravuje na dělení a kompletuje potřebné opravy. Posledním stadiem je buď mitóza či meióza a rozdělení buňky (cytokineze).
Cytokineze = oddělení cytoplasmy dvou dceřinných buněk (které po telofázi mají již samostatné jádro, ale jejich cytoplasma je ještě spojená). (pozn. překl.)
Je-li DNA buňky poškozena, p53 běžně zastaví růst buňky ve stadiu G-1 a odstartuje opravný proces. Genu p53 se říká „anděl strážný“ genomu, protože chrání tělo před mutacemi tak, že je opraví nebo (nejdou-li opravit) tak, že příslušnou buňku zničí a tak zabrání tomu, aby mutace byla předána potomstvu (Marx, 1994b). Bílkovina p53 to činí tak, že monitoruje kontrolní stanoviště buněčného cyklu G-1 a zastaví buněčný cyklus vyprodukováním proteinů, již přímo či nepřímo odstartují opravnou mašinérii DNA. Bílkovina p53 též spolupracuje přímo s reparačními proteiny.
Například poškození DNA způsobené ionizujícím zářením a jistými chemickými mutageny či ultrafialovým zářením (jež způsobují řetězcové zlomy DNA) může odstartovat akumulaci p53, která může zastavit cyklus buněčného růstu G-1. To umožní buňce opravit škodu na DNA před replikací; nebo, nemůže-li to být opraveno či selže-li oprava, p53 spustí sebedestrukční mechanizmus určený ke zničení defektní buňky apoptózou (Marx, 1994b; Sancar, 1994).
P53 kontroluje buněčný cyklus částečně také tím, že kóduje transkripční faktor, který aktivuje gen p21 k tomu, aby produkoval bílkovinu p21. Protein p21 pak blokuje buněčný cyklus zamezením všem komplexům cyklinu-Cdk v rozvoji, než je buňka opravena. Protein p21 také brání syntéze dlouhých úseků DNA, ale nebude blokovat výrobu kratších segmentů, které jsou třeba k opravě (Marx, 1994b). p21 se též účastní potlačování transkripce různých promotorů, jež nemají vazná místa pro p53, a dokonce brání aktivitě helikázy (proteinů rozvinujících DNA) a tak blokuje replikaci DNA.
Cyklin = regulační protein, který je nutný pro aktivaci některých typů proteinkináz. Uplatňuje se při řízení přechodů mezi jednotlivými fázemi buněčného cyklu.
Promotor = část DNA v blízkosti genu, která se podílí na regulaci jeho exprese. (pozn. překl.)
O p53 je známo více než o kterémkoli jiném proteinu k buněčné kontrole, zčásti proto, že jeho velká důležitost vedla k enormnímu zájmu vědců o tento gen v posledním desetiletí (Friedberg et al., 1995). Gen p53, a podobně mnoho jiných genů, které kontrolují buněčný cyklus, jsou geny pleiotropní; důsledkem je, že poškození genu p53 postihuje několik biochemických pochodů. Protein produkovaný tímto genem funguje také v udržování genomické plasticity a buněčné integrity. Váže se na různé opravné faktory transkripce, včetně XPD (Rad 3) a XPB, a účastní se opravy řetězce DNA přes jeho terminální doménu C (Wang et al., 1995).
Pleiotropie = vícečetný fenotypový projev funkce jednoho genu (jeden gen řídí současně fenotyp několika různých znaků).
Doména = část molekuly bílkoviny s určitou vlastností, funkcí apod. (pozn. překl.)
Apoptóza
Nemůže-li být buňka opravena, pošle ji p53 do komplexně naprogramovaného cyklu zničení zvaného apoptóza (Culotta a Koshland, 1993). Jedním z prostředků, jež buňka užívá, aby došla k apoptotické destrukci, je to, že nařídí lyzozomům, aby fungovaly jako „sebevražedné balíčky“tak, že jejich membrána praskne. Jejich obsah pak vyteče do cytosolu a způsobí strávení buňky zevnitř. Vyskytnou-li se mutace DNA, molekula p53 normálně opraví či zničí buňku dříve, než mutace způsobí problémy. Dojde-li k mutaci, jež znehodnotí p53, poškozené buňky nebudou zničeny a budou tedy schopny se reprodukovat a předat mutaci dále svému potomstvu. I v tomto případě sotva kdy dojde k mutacím přínosným, a dojde-li k expresi mutace, důsledkem je obyčejně nemoc (jako třeba rakovina).
Lyzozom = buněčná organela obsahující enzymy, které jsou schopny rozkládat („trávit“) pohlcený obsah, v některých případech části vlastní buňky.
Cytosol = termín užívaný v biochemii pro cytoplasmu, resp. homogenní, nestrukturovanou hmotu uvnitř buňky mimo organely a cytoskelet. (pozn. překl.)
Vypěstování kmene geneticky změněných myší, které neprodukovaly protein p53, pomohlo vědcům pochopit, jak tento mechanizmus funguje. V jedné studii se všechny myši narodily zdánlivě perfektně normální, ale po několika týdnech všechny myši bez proteinu p53 měly nádory – a za šest měsíců byly všechny mrtvé či umíraly na nádor. Jiné studie roku 1994 zjistily, že z 6,5 miliónu lidí s diagnózou rakoviny měla přinejmenším plná polovina mutace p53 (Modrich, 1994).
Mutace na p53 v gametě je předávána budoucím generacím zárodečnými buňkami, zatímco mutace vzniklé v buňkách somatických mohou vyvolat rakovinu či jinou nemoc. Osoby, které zdědí tento stav (zvaný Li-Fraumeniho syndrom) jsou obvykle v dětství zdravé, ale ve věku 30 let jich více než polovina rozvine jeden či více nádorů včetně nádorů mozku, osteosarkomu, leukémie a rakoviny prsu. Zdědění defektního genu p53 vždy způsobí rakovinu tehdy, když je oběť ještě mladá (často dříve, než se reprodukuje), a přes 90 procent takto postižených umírá na rakovinu před dosažením 70 let věku (Malkin et al., 1990). Bez ohledu na příčinu nádoru se p53 jeví jako gen fungující jako strážce. Rakovina se tedy často nerozvine, pokud není zajišťovací mechanizmus p53 poškozen. Jisté chemikálie v tabáku například ničí gen p53 a/nebo bílkovinný opravný proces, což je příčinou toho, že holdování všem typům tabáku je hlavní příčinou široké škály rakovin (Service, 1994).
Li-Fraumeniho syndrom = syndrom charakterizovaný výskytem zhoubných nádorů již od dětství. Příčinou je mutace tumor-supresorového genu p53.
Obranný systém těla proti mutacím může často pomoci zabránit zmutovaným genům způsobit škodu, i když je jeden z tumor-supresorových systémů buňky poškozen. Například velkou sadou opravných systémů je rodina proteinů k obraně vůči tepelnému šoku, třeba hsp 90 (Ridley, 2001). Tyhle bílkoviny pomáhají chránit buňku proti škodě, kterou může způsobit horko či jiný stres. Všechny tyhle obranné mechanizmy zajišťují, aby mutace zpravidla nemohly činit problémy v buňkách vystavených mutagenům. Je to jako trojí sada brzd; pokud první sada selže, může fungovat druhá či třetí. Stvořitel věděl, jak by se mohl buněčný mechanizmus poškodit, a tak vestavěl do buněk tyto komplexní mechanizmy k opravě či zničení zmutovaných buněk, aby se zabránilo makroevolučním změnám a nemocem.
Oprava pyrimidinového dimeru
Opravena může být dokonce i mutace pyrimidinových dimerů DNA (kde se dvě sousední thyminové či cytosinové báze nevhodně spojí). Takové dimerové mutace zaviňuje převážně ultrafialové světlo a jsou dosti běžné v buňkách pokožky. Jedna z opravných metod je označována jako oprava světlem čili oprava fotoreaktivací, protože proces opravy iniciuje viditelné světlo. Viditelné světlo aktivuje enzym, který rozruší pyrimidinové dimerové vazby, a tak opraví mutaci (Black, 1999). Jsou-li bakteriální kultury ozářeny ultrafialovým světlem, aby se u nich vyvolaly umělé mutace, musí se skladovat v temnu, aby se tak zabránilo mechanizmům opravujícím buňky ve zničení většiny těchto nových mutací, tedy mechanizmům závislým na světle.
Jiný typ opravy zvaný oprava temná vyžaduje několik reakcí kontrolovaných enzymy k odstartování potřebného opravného procesu (Black, 1999). Přesněji, restrikční endonukleáza přetne poškozené pyrimidinové dimery v přesném místě, a exonukleáza odstraní dimery i přilehlé nukleotidy (Jorde et al., 1997). Pak polymeráza opraví celý úsek s použitím správně postaveného komplementárního řetězce DNA jako vzorového templátu. Když polymeráza DNA syntetizuje novou DNA k nahrazení defektního úseku, třetí enzym znovu spojí staré a nové úseky. Když jsou napojeny, ligáza DNA opraví postranní strukturu nukleotidu. Opravné enzymy užité v tomto opravném systému kóduje nejméně sedm různých genů. Poprvé byl tento typ opravy identifikován u E.coli a další výzkum zjistil, že podobné opravné mechanizmy fungují u mnoha zvířat i lidí (Rosenfeld, 1983).
Restrikční endonukleáza = bakteriální enzym, který štěpí DNA v místě přesně určeném pořadím bází. Bakterie jej využívají k obraně proti virům.
Exonukleáza = enzym, který odštěpuje nukleotidy na koncích polynukleotidových řetězců.
Ligázy (syntetázy) = enzymy katalyzující vznik energeticky bohatých vazeb při současné hydrolýze ATP. (pozn. překl.)
Mutace v kterémkoli z těchto genů opravujících dimery může vést k defektu opravného mechanizmu a způsobit nemoci jako třeba xeroderma pigmentosum čili kožní rakovina. U pacientů s xeroderma pigmentosum začíná před 10. rokem věku rozsáhlá pihovatost následovaná kožními nádory, většinou na partiích těla vystavených slunci. Pak se rozvine závažné kožní zhoubné bujení vedoucí k časné smrti kolem 20. roku věku. Vyhýbání se všem zdrojům ultrafialového světla může omezit výskyt nádorů, ale nemůže trvale zabránit rozvoji rakoviny.
Oprava zlomeného chromozomu
Zlomení chromozomu, jež se vyskytne buď během meiózy či mitózy, je opravováno mechanizmem, který obvykle fixuje zlom „perfektně beze škody“ (Jorde, 1997) na dceřinné buňky. Tyto zlomy může způsobit spousta klastogenů (chemikálie nebo energie, jež lámou chromozomy), včetně ionizujícího záření či dokonce virových infekcí. Špatná oprava na chromozomu způsobí, či přispívá k rozvoji, mnoha nemocí včetně ataxie-telangiektázie, Bloomova syndromu, Fanconiho anemie a jiných (Bakkenist a Kastan, 2003).
Meióza = typ buněčného dělení, při němž z jedné buňky vznikají dvě dceřinné buňky, které mají jen polovinu (tj. 23) základního počtu chromozomů, z každého páru jeden náhodně vybraný. Uplatňuje se při vzniku pohlavních buněk (gamet) – vajíčka a spermie.
Mitóza = buněčné dělení, při němž z jedné buňky vznikají dvě buňky dceřinné, které mají zcela stejnou dědičnou výbavu.
Ataxie-telangiektázie je vzácná, ale fatální dětská nemoc, jež nepříznivě ovlivňuje jak neurologické tak imunologické funkce (Savitsky, 1995). U genu, somatického genu, který řídí opravu dvouřetězcového zlomu DNA, musí dojít k mutacím v obou kopiích, aby nemoc propukla (Bakkenist a Kastan, 2003). Asi 2 milióny Američanů jsou nositeli poškozeného genu, který způsobuje ataxii-telangiektázii.
Existuje mnoho dalších opravných systémů
Další nedávno objevený opravný mechanizmus, nonsense-mediovaná opravná cesta rozpadu mRNA, opravuje mRNA dříve, než může být použita k syntéze bílkoviny (Chin, 2001). Jakákoli mutace, jež působí přeskočení exonu, vyústí do přemístění mRNA do nonsense-mediované opravné cesty rozpadu mRNA k opravě (Liu et al., 2001). Nebo, pokud defekt v mRNA ústí do poškozeného terminačního kodonu, ribozomy nemohou být uvolněny a všechny ribozomy za tím uvíznou. Tento problém je detekován specifickou strukturou RNA zvanou tmRNA, jež se naváže blízko defektní mRNA.
mutace nonsense = substituční mutace bodová, při které vzniká z jiného kodonu kodon terminační. Taková mutace způsobí předčasné ukončení translace příslušné bílkoviny a tím ztrátu nebo poškození její funkce.
exon = část genu eukaryontů, která obsahuje vlastní dědičnou informaci.
nesmyslný kodon = kodon, který nekóduje žádnou aminokyselinu. Tímto kodonem je signalizováno ukončení translace daného polypeptidového řetězce, proto je označován také jako kodon terminační. (pozn. překl.)
Tento krok přidá specifickou sekci mRNA k defektní mRNA, která umožní, aby proces syntézy proteinů pokračoval, než ribozom dosáhne nového terminačního kodonu přidaného tmRNA. Uvolňovací faktor pak rozebere ribozom, což umožní ribozomům, které byly nakupeny, ukončit translaci řetězce mRNA. Bílkovina vyrobená touto modifikovanou mRNA má zvláštní sadu aminokyselin, která musí být odstřižena – krok, který učiní enzym zvaný koncová specifická proteáza (Silber et al., 1992; Beebe, 2000; Pallen a Wren, 1997).
proteazom = intracelulární proteinový komplex s proteolytickou aktivitou, který slouží k likvidaci přebytečných, afunkčních a defektních proteinů, nedokončených a špatně sestavených proteinových struktur. Je tvořen mnoha proteázami uspořádanými do válcovité struktury. (pozn. překl.)
Existují ještě další opravné systémy, jimž ještě plně nerozumíme. Během vývoje zvířete organizmus nějak prostě „ví“, kolik a jaké části těla jsou součástí plánu a kde se má ta která část nacházet. Tento systém může v podstatě anulovat chybu v instrukcích DNA, aby zajistil řádný vývoj (Ridley, 2001). Buňky mají dokonce „mechanizmy vnitřní péče, jež fixují další podíl chyb, jež prošly korekturami i filtry opravných enzymů“ (Ridley, 2001, str. 96).
Tento systém opravuje také škodu způsobenou reliktním zářením, volnými radikály i jinými zdroji. Současný odhad je ten, že každá buňka v těle přijme asi 10 000 zásahů denně jen od volných radikálů (což znamená, že každý řetězec DNA je vystaven asi 5000 zásahům denně) (Autry, 2003). To se rovná asi 630 kvadrilionům zásahů volnými radikály na každou osobu každý den.
Je jasné, že bez tohoto opravného systému by život vyšší než bakteriální nemohl přežít. Ačkoli o nich v tomto článku nepíšeme, všechny tyhle opravné systémy jsou „nezjednodušitelně složité“ (Behe, 1996) – což znamená, že žádná složka systému nemůže fungovat nezávisle na ostatních. Behe (1996) tvrdí, že neodarwinovská evoluce nemůže vysvětlit, jak vznikly takové provázané biologické systémy. Dodejme jen, že některé z těchto opravných systémů jsou nesmírně složité, a přes intenzivní studium jim stále ještě plně nerozumíme.
Opravný proces odporuje evoluci
Opravný proces zajišťuje extrémně nízký podíl mutací, který působí proti makroevoluci, protože mutace „jsou pro evoluci zásadní“ a koneckonců „veškerá genetická pestrost“ se zrodila z náhodných změn v sekvencích bází DNA, které představují surovinu pro evoluční proces (Audesirk a Audesirk, 1999, str. 167). Tento závěr implikuje myšlenku, že zvýšení podílu mutací bude přínosné, protože více mutací zárodečných buněk produkuje více pestrosti, a tato variabilita poskytuje surovinu, která umožňuje, aby probíhala evoluce.
Ačkoli darwinisté tvrdí, že mutace jsou původním zdrojem veškeré genetické pestrosti, všechna informovaná společenstva si dala mravenčí práci, aby udělala cokoli možného, jen aby omezila podíl mutací ve svém společenstvu na minimum. Rozsáhlý výzkum od přelomu 19. a 20. století dovedl vědce k závěru, že škoda z mutací daleko převyšuje jakékoli hypotetické přínosy, jež mutace mohou organizmu přinést (Bergman, 1995). A vskutku, jasně přínosná mutace, která by vyústila v získání „genetické informace“, nebyla nikdy dokumentována (Rust, 1992).
Ještě není známo, jak účinný je systém oprav mutací pro různé formy života – je možné, že je méně účinný u forem nižších (Selby, 1998). U těchto organizmů na nižší úrovni (jako třeba bakterií) nejsou mutace vždy opraveny tak účinně jako u většiny organizmů ostatních, a dokonce přispívají ke genetické proměnlivosti populace (Anderson, 2003). Některé mutace u bakterií mohou zřejmě vyprodukovat proteiny, jež umožní replikaci mimo dvouřetězcové zlomy, což je cestou k více mutacím v určitých genech a menší věrnosti replikace (podobný proces u vyšších organizmů iniciuje protein hsp 90). Tento zvýšený výskyt a udržení mutantů v bakteriální populaci je pokládán za mechanizmus přežívání, který umožňuje přinejmenším některým variantám přežít v nepřátelském prostředí (Anderson, 2003).
Darwinisté tvrdí, že se vyskytuje dost mutací, které nejsou opraveny, na to, aby byla umožněna evoluce. Některá nezhoubná pestrost nalézaná v životě je bezpochyby dílem mutací. Vycházíme-li však z toho, jak důležité jsou mutace pro naturalizmus, zdálo by se, že evoluce si vybírá jakékoli mechanizmy, jež by zvýšily podíl přínosných mutací.
Podle neodarwinizmu platí, že čím více se těchto mutací vyskytne, tím větší je naděje na produkci struktur, jež pomáhají zlepšovat poměry přežití i reprodukční schopnosti zvířete a tedy i evoluci. Mechanizmus, který omezí poměr mutací, by bránil produkci variací, která je ale jediným zdrojem makroevoluce. Jistěže by mutace také zvyšovaly sklon ke změnám nepříznivým, ale většina z nich by byla přírodním výběrem dosti rychle vyloučena. Takže z dlouhodobého pohledu by tyhle mutace nebyly důležité.
Nejdůležitějším faktorem v chodu makroevoluce je výskyt genetických mutací, které jsou výhodné, ale tyto změny jsou většinou možné pouze tehdy, vyskytne-li se mutací hodně – a darwinisté učí, že v dlouhodobém horizontu platí, že čím více mutací se vyskytne, tím větší je šance na vyprodukování mutace pozitivní. Z toho vyplývá, uzavírají, že mutace negativní jsou nutnou, ale malou cenou placenou za zlepšování.
Naopak model kreační postuluje, že genom byl původně perfektní, stejně jako ona spousta mechanizmů vyvinutých k produkci kontrolované genetické pestrosti, jako třeba genetický crossing-over během meiózy i sexuální reprodukce. Náhodné genetické změny by prakticky vždy narušily originální plán genomu, a tak ony mutace, jež jsou realizovány, by téměř vždy byly zhoubné (Marx, 1994a, 1994b). Existence komplexní „pečlivé mašinérie“ k zajištění vysoké věrnosti replikace DNA je plně v souladu s kreačním modelem. Opravný systém byl nejpodrobněji studován u bakterií, ale užitý mechanizmus je podobný u mnoha jiných organizmů včetně savců (Aboussekhra et al., 1995).
Crossing-over = překřížení odpovídajících částí chromatid analogických chromozomů během meiózy. Při c.o. může dojít k výměně příslušných částí chromatid s následnou novou kombinací dědičných vlastností na jednom chromozomu (rekombinace). Tento proces umožňuje vznik nových kombinací vlastností u potomků. (pozn. překl.)
Zdá se nám též, že evoluce vybírá proti vysoce efektivním opravným mechanizmům, protože odporují procesu, který umožňuje, aby makroevoluce existovala. Bez dostatečného množství genetické suroviny nemůže výběr působit tak, aby zlepšoval druh. Evolucionistické oponují, že tolik chyb najednou by bylo kontraproduktivní. McVean a Hurst (1997) tvrdí, že poměr mutací není nula díky výměnnému obchodu mezi „přínosy z redukce poměru zhoubných mutací, a náklady, jež vyžaduje stupňující se přesnost přepisu (náklady na čas a energii na korektury). Jinými slovy, možná, že existuje fyziologický limit na stupeň přesnosti v replikaci DNA“ (str. 388).
Avšak malé změny genomu jsou bezúčelné, a jedině velké genetické změny několika genů mohou produkovat dostatečnou inovaci k vytvoření komplexních nových struktur, které budou fungovat jako úplný, integrovaný celek. Kontrola více než 15 miliónů položek v literatuře nezjistila ani jediný článek, který by se snažil vysvětlit evoluci jakéhokoli mechanizmu k opravě mutace, ani článek, který by zkoušel přinést důkazy pro teorii, jak se opravné mechanizmy mohly vyvinout. Tento závěr potvrzuje zjištění Beheho (1996), že existuje úplné informační vakuum pro biochemickou evoluci téměř všech buněčných struktur, systémů i bílkovin.
Souhrn
Evoluční naturalizmus učí, že jediným zdrojem všech rysů produkovaných geneticky jsou mutace, které probíhají v linii zárodečných buněk. Takže podle této teorie je lidský genom tvořen hlavně mutacemi, které příroda vybrala, protože poskytovaly výhodu v boji o přežití v daném prostředí. Hlavním problémem tohoto postulátu je, že téměř všechny změny v kódujících genech, exonech, a prakticky také v mnoha dalších párech bází, budou opraveny opravným genovým systémem či jim bude zabráněno se projevit. A dále, pokud tento opravný systém řádně nefunguje, eventuální výsledek je mutace, která způsobí biologické problémy, nikoli pokrok (Hanawalt, 1994; Modrich, 1994). Takže genové mutace v exonech či jiné genetické poruchy jsou, jako celek, jasně regresívní a ne přínosné či progresívní.
Pokud nemohou být mutace účinně opraveny, dochází obvykle k vážnému či smrtelnému poškození zdraví. Nyní víme, že špatný opravný systém je původcem mnoha nemocí, jako třeba rakoviny. Dokonce i bakterie, jež vyrábějí „přínosnější“ mutace než vyšší organizmy, přece jen disponují komplexními opravnými systémy k opravám DNA. Bez takových opravných systémů by mutace brzy vyhubily všechen život, který by se vyvinul. Bez opravných systémů k vyloučení mutací by pouhé UV světlo rychle zničilo veškeré živočichy na Zemi. Všechny tyhle opravné mechanizmy jsou hlavními překážkami exprese genových chyb, jež údajně poskytují surovinu pro makroevoluci. Bez těchto komplexních opravných mechanizmů by organizmy postihl rychlý rozpad v důsledku mutací. Proto nedávný objev mnoha systémů opravujících mutace DNA je dalším vědeckým kamenem úrazu pro darwinovskou makroevoluci.
Poděkování
Chtěl bych poděkovat Susan Loeffelové, M.D., Davidu A. Demickovi, M.D., Waynu Frairovi, Ph.D., a Cliffordu Lillovi, M.A., a Bertu Thompsonovi, Ph.D., za jejich přínosné připomínky k dřívější verzi tohoto článku.
Odkazy
- Aboussekhra, A. M. Biggerstaff, M.K. Shivji, J.A. Vilpo, V. Moncollin, V.N. Podust, M. Protic, U. Hubscher, J.M. Egly, and R.D. Wood. 1995. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 80:859–868.
- Aldhous, P. 1995. DNA ‘fix’ offers safer cancer therapy. New Scientist 146(1972):16.
- Anderson, K.L. 2003. The complex world of gastrointestinal bacteria. Canadian Journal of Animal Science 83:409–427.
- Audesirk, G., and T. Audesirk. 2000. Life on Earth. Prentice-Hall, Upper Saddle River, NJ.
- Autry, D. 2003. Perilous peroxides. Custom Nutrition News 1(5):1–2.
- Bakkenist, C. J., and M.B. Kastan. 2003. DNA Damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 421:499–505.
- Bartek, J. and J. Lukas. 2003. DNA repair damage alert. Nature 421:486–587.
- Beebe, K.D., J. Shin, J. Peng, C. Chaudhury, J. Khera, and D. Pei. 2000. Substrate recognition through a PDZ domain in
tail-specific protease. Biochemistry 39:3149–3155. - Behe, M. 1996. Darwin’s black box. The Free Press, New York, NY.
- Bergman, J. 1995. Mutations and evolution. TJ Journal 9(2):146–154.
- Black, J.G. 1999. Microbiology; principles and explorations. Prentice-Hall, Englewood Cliffs, NJ.
- Chin, G. 2001. The dangers of nonsense. Science 291:211.
- Culotta, E., and D.J. Koshland. 1993. P53 Sweeps through cancer research. Science 262:1961–1958.
–—–—–. 1994. DNA repair works its way to the top. Science 266:1926–1929.
Eldredge, N. 1982. The monkey business. Washington Square Press, NewYork. - Friedberg, E.C. 2003. DNA damage and repair. Nature 421:536–440.
- Friedberg, E.C., G.C. Walker, and W. Siede. 1995. DNA repair and mutagenesis. AMS Press, Washington, D.C.
- Hanawalt, P.C. 1994. Transcription-coupled repair and human disease. Science 266:1957–1958.
- Harwood, J., and M. Meuth. 1995. Deletion mapping of highly conserved transcribed sequence downstream from APRT locus. Somatic Cell and Molecular Genetics 21(3):151–160.
- Jorde, L., J.Carey, and R.White. 1997. Medical Genetics. Mosby, St. Louis, MO.
- Kolodner, R. 2000. Guarding against mutation. Nature 407:687– 690.
- Liu, H.-X., L. Cartegni, M.Q. Zhang, A.R. Krainer. 2001. A mechanism for exon skipping caused by nonsense or missence mutations in BRCA1 and other genes. Nature Genetics 27:55–58.
- Mader, S.S. 1993. Biology. Wm. C. Brown, Dubuque, IA.
–—–—–. 1998. Biology. Second Edition. WCB/McGraw Hill, Dubuque, IA. - Malkin, D., F.P. Li, L.C. Strong, J.F. Fraumeni, Jr., C.E. Nelson, D.H. Kim, J. Kassel, M.A. Gryka, F.Z. Bischoff, M.A. Tainsky, and S.H. Friend. 1990 Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 250:1233–1238.
- Marx, J. 1994a. DNA repair comes into its own. Science 266:728–730.
–—–—–. 1994b. New link found between p53 and DNA repair. Science 266:1321–1322. - McVean, G., and L. Hurst. 1997. Evidence for a selectively favorable reduction in the mutation rate of the X-chromosome. Nature 386:388–392.
- Modrich, P. 1994. Mismatch repair, genetic stability, and cancer. Science 266:1959–1960.
- Pallen, M.J., and B.W. Wren. 1997. The HtrA family of serine proteases. Molecular Microbiology 26:209–221.
- Puck, T.T., R. Johnson, P. Webb, and G. Yohrling. 1998. Mutation measurement in mammalian cells. IV: comparison of
gamma-ray and chemical mutagenesis. Somatic Cell and Molecular Genetics 24:1–11. - Puck, T., R. Johnson, and S. Rasumussen. 1997. A system for mutation measurement in mammalian cells: application to
Γ-irradiation. Proceedings of the National Academy of Sciences, USA 94:1218–1223. - Radman, M., and R. Wagner. 1988. The high fidelity of DNA duplication. Scientific American 259(2):40–46.
- Ridley, M. 2001. The cooperate gene: How Mendel’s demon explainsthe evolution of complex beings. The Free Press, New York.
- Ritter, P. 1996. Biochemistry: A foundation. Brooks/Cole, New York.
- Rosenfeld, A. 1983. Master molecule, heal thyself. NSF Mosaic Reader. Avery, Wayne, NJ.
- Rozen, S., H. Skaletsky, J.D. Marszalek, P.J. Minx, H.S. Cordum, R.H. Waterston, R.K. Wilson, and D.C. Page. 2003. Abundant
gene conversion between arms of palindromes in human and ape Y chromosomes. Nature 423:873–876. - Rust, P. 1992. How has life and its diversity been produced? Perspectives on Science and Christian Faith 44:2:80–84.
- Sancar, A. 1994. Mechanisms of DNA excision repair. Science 266:1954–1956.
- Savitsky, K., A. Bar-Shira, S. Gilad, G. Rotman, Y. Ziv, L. Vanagaite, D. Tagle, S. Smith, T. Uziel, S. Sfez, M. Ashkenazi, I.
Pecker, M. Frydman, R. Harnik, S. Patanjali, A. Simmons, G. Clines, A. Sartiel, R. Gatti, L. Chessa, O. Sanal, M. Lavin, N. - Jaspers, A. Taylor, C. Arlett, T. Miki, S. Weissman, M. Lovett, F. Collins, and Y. Shiloh. 1995. A single ataxia telangiectasia
gene with a product similar to PI-3 kinase. Science 268:1749–1753. - Service, R.F. 1994. Stalking the start of colon cancer. Science 263:1559–1560.
- Silber, K.R., K.C. Keiler, and R.T. Sauer. 1992. Tsp: A tail-specifi c protease that selectively degrades proteins with nonpolar C
termini. Proceedings of the National Academy of Sciences, USA, 89:295–299. - Skaletsky, H., T. Kuroda-Kawaguchi, P.J. Minx, H.S. Cordum, L.Hillier, L.G. Brown, S. Repping, T. Pyntikova, J. Ali, T. Bieri,
A. Chinwalla, A. Delehaunty, K. Delehaunty, H. Du, G. Fewell, L. Fulton, R. Fulton, T. Graves, S.-F. Hou, P. Latrielle, S. Leonard, E. Mardis, R. Maupin, J. McPherson, T. Miner, W. Nash, C. Nguyen, P. Ozersky, K. Pepin, S. Rock, T. Rohlfi ng,
K. Scott, B. Schultz, C. Strong, A. Tin-Wollam, S.-P. Yang, R.H. Waterson, R.K. Wilson, S. Rozen,, and D.C. Page. 2003.
The male-specifi c region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423:825–837. - Sutherland, B., and A. Woodhead. 1990. DNA Damage and Repair in Human Tissues. Plenum Press, New York, NY.
- Wang, X.W, H. Yeh, L. Schaeffer, R. Roy, V. Moncollin, J.M. Egly, Z. Wang, E.C. Freidberg, M.K. Evans, B.G. Taffe. 1995. p53
modulation of TFIIH-associated nucleotide excision repair activity. Nature Genetics 10:188–195. - Weinberg, R.A. 1995. The retinoblastoma protein and cell cycle control. Cell 81:323–330.
- Willard, H.F. 2003. Tales of the Y chromosome. Nature 423:810–813.
- Wood, R.D. and T. Lindahl. 1990. Xeroderma pigmentosum. A gene for tumour prevention. Nature 348:13–14.
- Yang, A.S., M.L. Gonzalgo, J.-M. Zingg, R.P. Millar, J.D. Buckley, and P.A. Jones. 1996. The rate of CpG mutation in alu repetitive elements within the p53 tumor suppressor gene in the primate germline. Journal of Molecular Biology 258:240–250