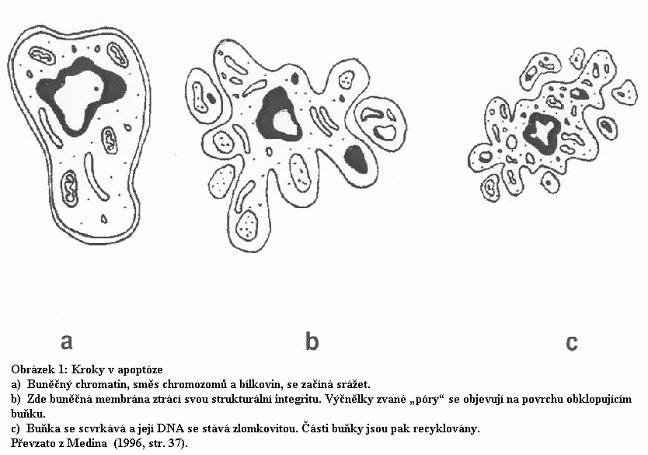
Původ
apoptózy: sobecké geny či inteligentní plán?
(Origins of Apoptosis: Selfish Genes or Intelligent Design?)
CRSQ, roč. 44 (zima 2008), č. 3, str. 204 – 211, přeložil M. T. – 5/2008
http://www.creationresearch.org/members/crsq/44/44_3/CRSQ_Winter_08_lo_res.pdf
Apoptóza je složitý biochemický mechanizmus, kdy se specifická buňka zničí za jistých podmínek, bez újmy pro okolní buňky. Přehled literatury odhaluje, že neodarwinisté nejsou schopni vysvětlit, jak se mechanizmus pro apoptózu mohl vyvinout procesem krok za krokem. Mechanizmus apoptózy reguluje buněčný život ku prospěchu organizmu jako celku a nikoli ku prospěchu pouhých genů, jak navrhoval Dawkins. Apoptóza podporuje koncepci neredukovatelné složitosti a inteligentního plánu.
Buňky mohou být eliminovány dvojím způsobem: nekrózou (smrt buňky způsobená zraněním či chorobou) a apoptózou (programová smrt buňky). Apoptóza je velmi důležitým mechanizmem užívaným všemi mnohobuněčnými organizmy k vyloučení nepotřebných či škodlivých buněk včetně rakovinných a prekancerózních buněk (Shi, 2002; Steller, 1995; Huang a Strasser, 2000 str. 839; Weinrauch a Zychlinsky, 1999). Apoptóza také hraje důležitou úlohu v normálním vývoji, ve fyziologické rovnováze (homeostáze) a v obměně buněk (Elmore, 2007; Ashkenazi a Dixit, 1998).
Počínaje embryonálním stadiem, buněčné dělení a apoptóza musí být v jemné rovnováze a synchronizaci, aby udržely zdraví a život. Například po opalování apoptóza zničí poškozené buňky, které by se mohly stát rakovinnými, čímž umožní jejich nahrazení. Apoptóza má za následek smrštění buněk, mitochondriální zhroucení, uvolňování cytochromu C, vývoj pórů na povrchu buňky, a recyklaci buněčných komponentů.
Cytochromy = hemoproteinové pigmenty s funkcí oxidoredukčních enzymů a různou nitrobuněčnou lokalizací. Mitochondriální cytochromy (a, a3, b, c) jsou součástí dýchacího řetězce. (pozn. překl.).
Neredukovatelná složitost je „Jediným systémem, složeným z několika spolupracujících částí, které přispívají k základní funkci systému, kde odstranění kterékoli části způsobí, že systém přestane účinně fungovat“ (Behe, 1996, str. 9). Příkladem neredukovatelné složitosti je fakt, že chemický prvek uhlík musí mít šest protonů. Odejmeme-li jeden proton, už neexistuje uhlík, ale vznikne bór. Inteligentní plán používá sadu kritérií ke zhodnocení fyzikálního světa, zda je za nějakou událost odpovědná inteligence. Běžným příkladem ze soudní praxe je fakt, že musí být určeno, zda mrtvé tělo zemřelo na nemoc, nehodu či plánovaným zásahem – specificky v právní terminologii sebevraždou nebo vraždou.
Dnes jsou známy tři cesty, které spouštějí apoptózu. První je zevní systém čili receptor smrti spouštěný signálem z vnějšku buňky (smrtelný ligand, který se váže na receptor smrti). Druhou je vnitřní systém spouštěný signálem z vnitřku buňky působený faktory zahrnujícími záření, jedy, a hypoxii. Poslední je perforinový systém způsobený cytotoxickými T buňkami. Tyto tři systémy spouštějí cestu exekuce čili cestu způsobující nakonec zničení buňky (Elmore, 2007).
Ligand = látka (molekula, atom), která je schopna se vázat na buněčný receptor (jinou molekulu).
Hypoxie = nedostatek kyslíku.
Perforin = protein podílející se na působení cytotoxických T lymfocytů; po degranulaci tvoří póry v likvidované buňce; je závislý na kalciu. (pozn. překl.).
Buňka prodělávající apoptózu vždy vykazuje jistou sadu charakteristických znaků, zahrnujících smrštění buňky a pyknózu, stažení buněčného jádra do kompaktní masy, jež účinně přijímá barvivo (Elmore, 2007). Vlastní apoptóza začíná, když proteázy známé jako spouštěcí kaspázy jsou aktivovány uvnitř buňky jako důsledek proteolytického procesu, který způsobí, že jiné proteázy pohltí specifické složky buňky (Hengartner, 1998).
Proteináza (též proteáza) = enzym rozkládající bílkoviny na menší části – peptidy.
Kaspázy (caspases) = skupina proteolytických enzymů obsahujících v aktivním centru cystein.
Proteolýza = rozložení bílkovin na menší části, tj. peptidy a aminokyseliny. (pozn. překl.)
Proteázy zvané kaspázy fungují při apoptóze tak, že řídí složité specializované molekuly, které rozpouštějí buněčný chromatin, ničí nukleoskeleton buňky, ničí enzymy, které zmnožují a opravují DNA, a aktivují CAD. CAD je zkratka tří enzymů, karbamoylfosfátsyntázy, transkarbamylázy aspartátu, a dihydroorotázy, které rozkrájejí DNA na malé, přibližně stejně velké části o 200 párů bází (Rupinder et al., 2007).
Karbamoylfosfát = reaktivní mezistupeň fungující jako přenašeč karbamoylové skupiny na jinou vhodnou molekulu.
Syntázy = lyázy, enzymy schopné spojit dvě sloučeniny kovalentní vazbou. Jde o triviální název pro lyázy, který zdůrazňuje „syntetický“ směr reakce.
Aspartát = sůl nebo ester kyseliny asparagové. (pozn. překl.)
Kaspázy též aktivují enzymy, které tráví cytoskeleton, ničí schopnost buňky srůstat s jinými buňkami, a nakonec rozlámou buňku na malé zlomky (Barinaga, 1998). Způsobují i přesun fosfolipidu z vnitřního povrchu membrány apoptotické buňky na její povrch, aby přilákaly fagocyty. Fakt, že „všechny známé kaspázy sdílejí podobné prostorové uspořádání na skupině vážící substrát“ naznačuje neredukovatelnou složitost, protože specifický komplexní plán je nutný pro to, aby systém fungoval. A v živém světě není známo žádné stupňování ve složitosti tohoto designu (Shi, 2002, str. 460). Všechny známé organizmy, u nichž probíhá apoptóza, vyžadují tento systém kaspáz ke své řádné funkci (Rupinder et al., 2007).
Druhá skupina proteáz účastnících se apoptózy zahrnuje enzym štěpící proteiny zvaný „interleukin I měnící proteázy podobné enzymům“, zkráceně proteázy ICE. Proteázy ICE pomáhají zničit buňku strávením základních proteinů a jistých strukturálních složek v genetickém materiálu buňky, aby zabránily v tom, aby se buňka opravila (Martin et al., 1995). Tento dvojí útok na buňku zaručuje jak její smrt tak její bezpečnou destrukci bez nepříznivého postižení buněk jiných.
ICE = interleukin I beta converting enzyme, kaspáza I (pozn. překl.).
Dalším krokem v apoptóze je proces, kdy buňka vysílá atraktanty, jako třeba fosfatidylserin (zvané signály „sněz mě“) z listu vnitřní buněčné membrány k listu vnější buněčné membrány, aby přilákala fagocyty. Normální interakce buňky s buňkou, jako třeba schopnost sloučit se s buňkou jinou, jsou též rychle ztráceny u buněk podstupujících apoptózu. Buňka se pak postupně stává kulatější, a její buněčná membrána se zvlní, tvoříc výčnělky, zduřeniny zvané póry (viz obrázek 1). Brzy se rozpadá a zbytky její membrány opouzdří zlomky buňky, které jsou v tomto stadiu známy jako apoptotická tělíska (Veggeberg, 1995). To omezuje množství uniklých apoptotických buněčných toxinů nebo zhoubného obsahu buňky (Elmore, 2007).
Fosfatidylserin = druh fosfolipidu, ve kterém je na zbytek fosforečné kyseliny estericky vázána aminokyselina serin.
Apoptotické tělísko = svraštělá buňka, která je posléze fagocytována, aniž dojde k úniku proteolytických enzymů a vzniku zánětlivé reakce. (pozn. překl.)
Buněčné zbytky vystaví antigeny, které způsobují, že jsou pozřeny parazitickými makrofágy, které žijí ve všech tkáních, a jinými místními parazitickými buňkami. Zlomky buňky jsou pak rozlámány lyzozomálními organelami a výživné látky buňky jsou recyklovány bez spuštění zánětlivé odpovědi typické pro nekrózu.
Veškerý apoptotický proces netrvá ani hodinu, což je jeden z důvodů, proč nebyl donedávna biology objeven (Raff, 1996).
Makrofág = buňka, která je schopna pohlcovat (fagocytovat) cizorodý materiál včetně mikroorganizmů. Plní i další významné úlohy v imunitě, produkuje cytokiny, zpracovává a prezentuje antigeny.
Lyzozom = buněčná organela obsahující enzymy, které jsou schopny rozkládat („trávit“) pohlcený obsah, ale v některých případech i části vlastní buňky. (pozn. překl.)
Existence apoptózy byla předpokládána od úsvitu teorie buňky, ale byla popsána teprve nedávno. Kdysi se myslelo, že smrt buňky nevratně ústí do záporných důsledků pro tělo, ale v 50. letech výzkum ukázal, že jisté buňky jsou systematicky eliminovány jako normální součást vývoje. Příklady zahrnují ocas pulce, zřasení mezi prsty u lidských embryí, a změnu prsní tkáně, když matka přestane kojit ( Duke et al., 1996). Apoptóza též organizuje mozek tak, že eliminuje neurony, které nemají správné zapojení s jinými nervovými buňkami. Apoptóza dokonce způsobuje ztrátu velkých skupin buněk, jako třeba opad korunních lístků květu. Tahle posledně jmenovaná role pro apoptózu je zdrojem termínu nyní používaného pro označení celého procesu (apoptosis je řecky „opad“). Termín apoptóza byl poprvé užit teprve roku 1972 (Elmore, 2007).
Průkopnický článek zveřejněný roku 1972 načrtl první důkazy pro apoptózu v prekancerózních buňkách (Kerr, Wyllie, a Currie, 1972). Bohužel, tento článek zůstal do velké míry nepovšimnut až do začátku 80.let. Práce Roberta Horvitze o Caenorhabditis elegans (často studovaný hlíst) byla také velmi důležitá pro pochopení apoptózy (Veggeberg, 1995).
K apoptóze dochází u jediné buňky a jejího genomu, ale prospívá celému organizmu. Všechny eukaryotické buňky obsahují všechny geny potřebné pro samozničení a učiní tak, pokud nejsou tyto geny poškozeny, jako při rakovině, či pokud buňka nedostane signály od jiných buněk, aby apoptózu blokovala (Raff, 1996). Udržování přesné rovnováhy mezi buněčným dělením a buněčnou smrtí prospívá organizmu v několika ohledech. Život na hraně buněčné sebevraždy zajišťuje, že všechny poškozené buňky jsou rychle zničeny a že jejich těla jsou účelně a účinně pohlcena buňkami sousedními (Adams a Cory, 1998; Wu, 1996).
Apoptóza je zvláště důležitá pro pochopení mnoha složitých buněčných procesů, jako třeba jak inzulinoidní růstový faktor I a jiné bílkoviny mohou kontrolovat a eliminovat vybrané buňky. Znalost apoptózy je třeba k pochopení toho, jak p53 a jiné velmi důležité tumorosupresivní geny fungují (Mercer et al., 2007). Tyhle tumorosupresivní geny regulují opravu poškozené DNA, ale je-li poškození příliš velké, geny způsobí, že apoptóza buňku zničí.
Růstové faktory = látky peptidového charakteru, které ovlivňují a regulují růst buněk. Určitý efekt jako růstové faktory mají i některé hormony, včetně např. inzulinu. (pozn. překl.)
Odchylná regulace apoptózy může dovolit buňkám s mutacemi (jako třeba rakovinným buňkám) pokračovat v dělení a následně taková odchylná regulace přispívá k progresi rakoviny a jiných nemocí. Špatné nastavení apoptózy může způsobit zničení buněk, které nejsou poškozeny jak během, tak i a po infarktu. Pokud jsou poškozené opravitelné buňky zničeny, plyne z toho zbytečné poškození srdce.
Špatné nastavení apoptózy může přispět i k dalším poruchám, jako jsou nádory a jisté autoimunitní choroby jako reumatoidní artritida (Rupinder et al., 2007; Barinaga, 1998). Dokonce i Alzheimerova, jakož i Parkinsonova a Huntingtonova nemoc jsou všechny považovány za nemoci, jichž se účastní stav, který přinutí specifický neuron spáchat sebevraždu předčasně (Fesik, 2000). Také se má za to, že apoptóza se využívá k vylaďování imunitního systému tak, že eliminuje buňky T, jež útočí na vlastní buňky člověka – krok nutný k rozvinutí normální autoimunity.
Náhodné zničení buňky, známé jako nekróza, je za normálních okolností nežádoucí. Ale apoptóza, pokud je řádně řízena, je přínosná. Slovo nekrotický vychází z řeckého slova znamenajícího zabít. Ke smrti buňky nekrózou dojde, pokud je buňka vážně zraněna fyzikálním či chemickým způsobem, jako třeba nedostatkem kyslíku. Při nekróze buňky většinou otečou a rozkládají se, přitom vypouštějí cytoplazmatický materiál, který spustí zánětlivou odpověď v mezibuněčné základní hmotě (Elmore, 2007). Havním rozdílem mezi nekrózou a apoptózou je fakt, že při nekrotické smrti je buňka pasivní obětí, avšak u apoptózy je buňka aktivním účastníkem, dokonce vynakládá vlastní energii na to, aby dosáhla vlastního zániku (Zamzami a Kroemer, 2001). Zda buňka zemře nekrózou nebo apoptózou, to závisí zčásti na faktorech zahrnujících signální specifika smrti buňky, fyziologické prostředí, stav tkáně a stadium vývoje buňky (Elmore, 2007, str. 496).
Mitochondrie hrají důležitou a ústřední roli ve zprostředkovávání vnitřního systému apoptózy (Hengartner, 1998a). U nekrotické smrti, mitochondrie a další buněčné organely jsou často prvními, které otečou a vyhřeznou, následovány rozkladem celé buňky. U apoptózy, místo otoku buňky, vnitřní mechanizmy jako třeba působení kaspáz, způsobí, že se buňka scvrkne. Jak se apoptózovaná buňka scvrkává, odtahuje se od buněk sousedních. Výsledkem je, že apoptotická buňka v normálním případě nespustí zánětlivou odpověď. Jádro se také dramaticky scvrkne u apoptózy, a chromatin se sráží do zřetelných pórů, jež pak putují k obalu jádra (Duke et al., 1996, str. 80).
Chromatin = hmota buněčného jádra tvořená DNA a bílkovinami, která je uspořádána do chromozomů. (pozn. překl.).
Výjimky z obvyklé eliminace buněk apoptózou se týkají několika málo buněčných typů, jako třeba těch, které tvoří oční čočky. U dospělého sestávají oční čočky především z mrtvých těl buněk. Čočka vzniká z buněk pomalu odumírajících, a během této doby je většina cytoplazmy postupně nahrazována krystalickou bílkovinou (Duke et al., 1996). Další příklady zahrnují buňky pokožky, které, jak zrají, nahrazují svůj obsah bílkovinou keratinem, získávají povlak odolný proti vodě, odumřou, a nakonec se odloupnou, aby byly nahrazeny buňkami vzlínajícími zpod nich.
Náležitosti apoptózy se mění také podle typu buňky. O apoptóze T-lymfocytů je známo více než o apoptóze většiny dalších typů buněk. T-buňky vznikají z předchůdců kostní dřeně, putují do brzlíku jako thymocyty, a pak se vyvinou ve specializované T-buňky, které vyvíjejí receptorové molekuly, jež umožňují zralým T-buňkám zjišťovat specifické antigeny (McColl et al., 2007; Gregory, 1995). Thymocyty, jež buď nejsou s to produkovat funkční receptory nebo produkují receptory, které odpovídají na buňky vlastního těla, jsou též zničeny apoptózou. Platí zejména, že thymocyty, které jsou ničeny, jsou ty, které se váží příliš silně na molekuly vyvinuté v brzlíku, což znamená, že mohou zasáhnout později zdravou tkáň a způsobit autoimunitní chorobu (Golstein et al., 1991). Defektivní apoptóza může umožnit některým z těchto autoreaktivních buněk přežít, což má za následek špatné fungování imunitního systému (Fesik, 2000). Nejlepšími příklady zde jsou různé autoimunitní choroby.
Apoptóza je spouštěna buď kontrolovaným nedostatkem jistých faktorů potřebných k přežití buňky, nebo mechanizmem uplatňujícím takzvaný receptor smrti jako třeba Fas, Apo 1, CD95 a mnoho dalších bílkovinných receptorů smrti. Tyhle bílkovinné receptory všechny patří do superrodiny genů s faktorem nádorové nekrózy (TNF), které jsou společně zvány ligandy smrti. Některé růstové receptory, jako třeba receptor pro faktor nervového růstu (NGF), také obsahují sféru smrti, která pomáhá regulovat růst a opravy (Ashkenazi a Dixit, 1998). Ligandy, které se váží na tyto receptory, aktivují všechny apoptózu a potud se zdá, že jsou všechny strukturálně podobné. Jak je běžné u mnoha bílkovin užívaných při apoptóze, nevykazují stupňování od jednoduchých k složitým, jak očekává neodarwinizmus.
Smrt buňky může být spuštěna vnějšími nebo vnitřními buněčnými signály, růstem, faktory přežívání, či dokonce konfliktními signály, které regulují dělení buněk (Fesik, 2000). Buňky, které napadne virus, také často spustí apoptózu. Apoptóza může být spuštěna řadou dalších mechanizmů, včetně p53 a jiných genů pro potlačení nádorů. Buňky s DNA poškozenou mutageny spouštějí produkci bílkoviny p53, která pak aktivuje sadu kroků, jež mohou zahrnovat apoptózu, a tak vést ke zničení buňky postižené mutací. Tahle řada mechanizmů, jež slouží k opravě (či zničení) buněk s mutacemi je velkým problémem pro neodarwinizmus. Kdyby opravy fungovaly spolehlivě, nemohlo by dojít k evoluci. Bez oprav by nemohl život, jak jej známe, existovat (Elmore, 2007).
Specifické signály, o nichž je známo, že spouštějí apoptózu, se mění podle typu buňky a dalších faktorů, jež ústí do výběrové eliminace specifických buněk (Huang a Strasser, 2000). Apoptóza může být odstartována také prostě jen působením času. Keratinocyty – druh buněk pokožky – podléhají stárnutí cestou apoptózy asi 21 dnů po tom, co začnou putovat ven směrem k povrchu pokožky.
Vnímavost a náchylnost buňky vůči apoptóze se mění podle několika faktorů. Sada bílkovinných molekul pevně reguluje apoptózu složitými způsoby; některé usnadňují spouštění, zatímco jiné ústí do útlumu (Korsmeyer, 1995). Rodina bílkovin zvaná Bcl-2 apoptózu tlumí, zatímco jiná rodina příbuzných bílkovin, zahrnujících bílkoviny Bad, Bak, Bok, Bik, a Bid, celá apoptózu podporuje (viz obrázek 2). Rovnováha podporujících a tlumících faktorů slouží tomu, aby bylo zajištěno, že apoptóza bude spuštěna pouze tehdy, kdy je třeba, a blokována tehdy, kdy třeba není (Zamzami a Kroemer, 2001).
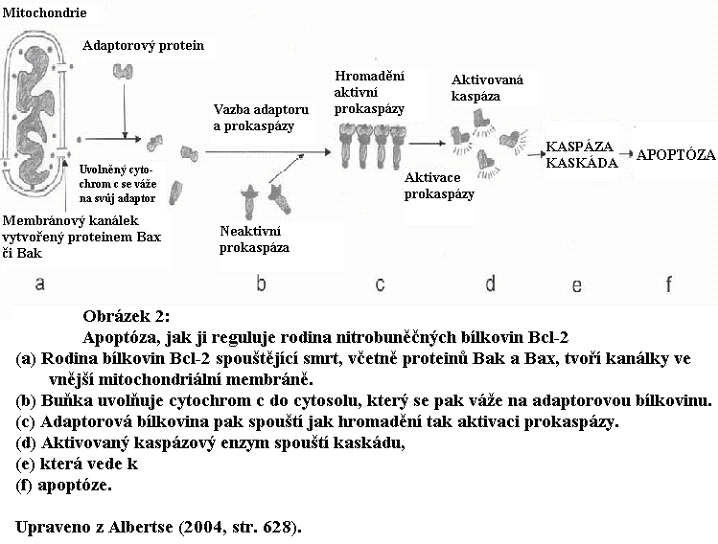
Cytochrom c = cytochrom, který je součástí dýchacího řetězce a podílí se na transportu elektronů v membráně mitochondrie.
Cytosol = termín užívaný v biochemii pro cytoplazmu, resp. homogenní, snad nestrukturovanou hmotu uvnitř buňky mimo organely a cytoskelet.
Kaskáda = systém složený z postupně se aktivujících složek, který umožňuje výsledné mnohonásobné zmnožení či zvýšení účinku. V organizmu funguje řada dějů na principu kaskády, např. nitrobuněčný přenos signálů. (pozn. překl.)
Řada funkcí proteinů zapojených do apoptózy je nyní aktivně zkoumána, a další jsou každým rokem objevovány. Lewin (1997, str. 1128) si všiml, že funkce jedné z nejstudovanějších rodin regulátorů apoptózy, Bcl-2, jsou stále „záhadou“, ale většina reguluje signály, jež vedou k aktivaci kaspáz (Huang a Strasser, 2000). Proteinová rodina Bcl-2 zahrnuje jak proapoptotické tak antiapoptotické molekuly, které hrají klíčovou roli v rozhodování o tom, zda buňka bude žít či nikoli (Gross et al., 1999). Bcl-2 má kotvu C-konce, kterou také nacházíme nejen na vnější jaderné, ale též na mitochondriální membráně i membráně endoplazmatického retikula – což naznačuje, že patrně kontroluje široké spektrum buněčných funkcí (Gross et al., 1999).
C – konec = označení pro karboxylový konec proteinu (tj. část bílkoviny, kde příslušný aminokyselinový zbytek je vázán svou aminoskupinou a jeho karboxylová skupina –COOH zůstává volná) – pozn. překl.
Smrt buňky může být blokována produkcí
vysokých hladin proteinu Bcl-2 tlumícího apoptózu (Hartwell a
Kastan, 1994; Hartwell a Weinert, 1989). U lymfocytů gen bcl-2
blokuje apoptózu, kdežto gen bax ji spouští. Normálně jsou
bílkoviny Bcl-
Dimer = chemická sloučenina složená ze dvou stejných (homodimer) či rozdílných (heterodimer) jednotek. (pozn. překl.)
Jisté normální buňky, které by
způsobily fatální následky, kdyby byly pro tělo ztraceny,
jako třeba buňky srdce, produkují relativně vysoké hladiny
bílkoviny Bcl-2. Je tedy menší pravděpodobnost, že budou
zničeny apoptózou, a větší pravděpodobnost, že budou
opraveny (Mercer et al., 2007). Pokud se stanou rakovinnými,
tyhle buňky mohou vytvářet agresivní nádory. Buňky
melanocyty produkují pigment melanin, který způsobuje
tmavnutí pokožky, a tím chrání škáru před poškozením
ultrafialovým zářením. Kdyby melanocyty hynuly příliš
snadno, ztráta ochranné funkce, která by následovala, by
způsobila, že další buňky by byly v mnohem větším riziku
poškození ultrafialovými paprsky. Ale melanocyty produkují
velké množství Bcl-
Buňky (jako třeba buňky srdečního svalu), které nejsou náchylné k poškození UV zářením, produkují velmi malé množství bílkoviny Bcl-2 tlumící apoptózu. Proto mají tendenci prodělávat zničení apoptózou dost často. Tento systém normálně chrání tyto buňky proti rakovině, ale nedostatek správné rovnováhy může též působit proti nejlepšímu zájmu těla (Mercer et al., 2007).
Apoptóza funguje podobně při virových infekcích, a to několika způsoby. Aby se mohl rozmnožit, musí virus potlačit schopnost buňky vyrábět všechny bílkoviny kromě těch potřebných k produkci dalších virů. Hostitelova vlastní syntéza bílkovin je tak blokována, což může spustit apoptózu, zabíjející jak buňku tak virus (Fesik, 2000). Toto normálně chrání buňku před širokým spektrem virů.
Aby tomu zabránil, virus Epstein-Barrové,
který způsobuje mononukleózu, a jisté další viry, disponuje
geny potlačujícími apoptózu, které kódují pseudo Bcl-2
protein, jenž je strukturálně podobný proteinu Bcl-2. Ten
může dokonce fungovat jako normální protein Bcl-
Rakovina je ztrátou normální kontroly nad buněčným dělením, rozrůzňováním, a dalšími buněčnými funkcemi. Je způsobena poškozením buněčné DNA. Aby se rakovina rozvinula, musí být mechanizmus apoptózy přerušen nebo odložen na tak dlouho, aby to umožnilo některým buňkám navršit dosti mutací tak, aby byly s to se nekontrolovatelně dělit, a nakonec metastázovat (Fesik, 2000). Výsledkem je produkce buněk, jež mají genetické mutace, a proto fungují nenormálně. Tyto rakovinné buňky neutrpěly dostatečnou škodu na to, aby vymřely. U rakovinných buněk genetické poškození obvykle vede k neschopnosti zahájit apoptózu, protože gen, který kóduje buď protein p53 či některý jiný gen pro potlačování nádorů, je zmutovaný nebo ochromený (Wang et al., 1995). Rakovinné buňky se pak mohou stát imunními vůči normálním spouštěčům apoptózy.
Znalost vztahu mezi apoptózou a rakovinou může usnadnit naše využívání apoptotických mechanizmů k diagnóze prekancerózních stavů. Může snad též pomáhat při určování místa rakoviny. Nejlepším příkladem je folikulární lymfom, způsobený přemístěním genu bcl-2 z chromozomu 14 na 18, což propůjčuje nesmrtelnost imunitnímu systému B buněk, jež se pak mohou nashromáždit do nebezpečných hladin podobných leukémii.
B lymfocyt (B buňka) = druh lymfocytu, který se podílí na humorální imunitě (tvorbě protilátek) a na některých dalších imunitních funkcích. (pozn. překl.)
Poškození způsobené kyslíkovými volnými radikály je hlavní příčinou mutace, která může pak vést k rakovině, stárnutí, šedým zákalům, ateroskleróze a dalším chorobám. Kyslíkové volné radikály mohou též spustit apoptózu, pokud poškození není dostatečné k tomu, aby zničilo buňku nekrózou. Strava bohatá na neutralizéry volných radikálů, jako třeba vitamíny A, C, E a selen, může omezit poškození způsobené volnými radikály.
Programová smrt buňky je způsobena mnohými z týchž genů, které kontrolují cyklus buněčného dělení (Maddika et al., 2007). Funkční přístup ke studiu programové smrti buňky zahrnul viry, které ochromíme v jejich funkci patogenů odstraněním genů způsobujících chorobu. Geny, které pak fungují jako bojovníci proti chorobě, jsou pak naroubovány do genomu viru. Virus je pak schopen naroubovat své nové antivirové geny do genomu DNA buněk, které napadne.
Příklad toho, jak je tento přístup využíván k zabíjení rakovinných buněk, zahrnuje naočkování nádorů geny obsahujícími modifikovaný virus oparu, které spouští apoptózu. Virové geny, jež mohou být takto vpraveny do genomu buňky, zahrnují „sebevražedné geny“, způsobující, že tyto buňky jsou vysoce citlivé vůči lékům jako je ganciklovir, který se užívá k léčbě oparu (Hartwell a Kastan, 1994). Tento přístup nakazí buňky virem, který jim způsobí pseudo-nemoc. Poté jsou léky účinné v zabíjení „pseudo-nemocných“ buněk užity k zabití nakažených buněk. Léčení lidé jsou též naočkováni stimulantem, který je užit k zesílení účinku „sebevražedného genu“. Problémem, který ještě musí být vyřešen, je fakt, že modifikovaný virus oparu stále ještě spouští imunitní systém, který pak napadá modifikovaný virus a překáží účinku sebevražedného genu. Je třeba vyvinout techniky k určení toho, jak by geny virů mohly zůstat skryty či jak by imunitnímu systému mohlo být zabráněno v rozpoznávání a ničení těchto virů. Viry mohou též být užity jako taxi dopravující geny do buňky k nahrazení či „fixování“ poškozených genů, které vedou k rakovině.
Jen malá část znalostí o biologickém světě známých dnes byla k dispozici v 19.století, když William Paley psal o svém názoru na design spatřovaný v kapesních hodinkách. To, co jsme se dověděli od té doby, podpořilo výmluvně Paleyovu tezi o tom, že plán je jasným důkazem pro Plánovače. O úrovni složitosti v buňce se nyní ví, že přesahuje úroveň složitosti existující nad buněčnou úrovní. Apoptóza je pouze jedním z mnoha tisíc složitých buněčných systémů, které nyní zkoumáme. Udělal jsem si počítačovou rešerši z více než 15 miliónů záznamů ze dvou databází a objevil téměř 70 000 článků o apoptóze. Ale jen čtyři z těchto 70 000 se vzdáleně týkaly evoluce apoptózy. Žádný z nich neobsahoval empirické důkazy, které by odporovaly závěrům Beheho (1996) či Paleye (Blackstone a Green, 1999; Wiens et al., 2000; Aravinch et al., 1999).
Jeden ze tří článků, které se zmiňovaly o evoluci, byl vysoce spekulativní diskusí o roli mítochondriálního cytochromu c v apoptóze (Blackstone a Green, 1999). Autoři spekulovali, že apoptóza u složitějších zvířat je snad zbytkem evolučních konfliktů mezi předpokládanou endosymbiózou mitochondriální organely a jejího buněčného hostitele. Ale endosymbióza, víra, že organely v metazoických buňkách vznikly, když buňky předků zachytily mikroby, je sama o sobě myšlenkou, která trpí několika velkými problémy (Bergman, 1998).
V dalším článku, Wiens et al. (2000) hodnotili srovnávání sekvencí jistých organických molekul účastnících se apoptózy, jako třeba superrodiny Bcl-2. Zjistili velkou podobnost mezi jistými metazoickými organizmy a obratlovci. Třetí autor se pokoušel vysvětlit, jak se může neodarwinizmus vypořádat se dvěma opačnými řešeními problému přežití virů. Někdy viry apoptózu tlumí, a jindy mohou viry apoptózu stimulovat (Krakauer a Payne, 1997). Žádný z těchto článků neměl jako hlavní téma makroevoluční původ apoptózy.
Poslední objevený článek (Kroemer, 1997) byl prvním vážným pokusem vysvětlit evoluci apoptózy. Kroemer užil endosymbiotickou teorii k pokusu vysvětlit, jak se mohla apoptóza vyvinout. Jeho diskuse účinně ilustruje obtíže v evoluci apoptózy. Také se pokouší spekulovat o tom, jaký je evoluční vztah nekrózy k apoptóze, ale dochází pouze ke zdůraznění oněch mnoha rozdílů mezi těmito dvěma systémy. S málo výjimkami (jako třeba blastomery nebo buňky očních čoček), všechny buňky u vícebuněčných živočichů jsou určeny k podstoupení apoptózy, pokud není zablokována signály z buněk jiných (Raff, 1996). Tento fakt je užit jako důkaz,že apoptóza se vyvinula velmi záhy v jednobuněčných orgánech. Problémem se všemi evolučními scénáři je fakt, že apoptóza by prospívala pouze mnohobuněčným organizmům, protože u jednobuněčného tvora by ukončila genetickou linii.
Blastomery = buňky vzniklé několika prvními děleními (rýhováním) zygoty. (pozn. překl.)
Pokud je známo, apoptóza funguje velmi podobným způsobem ve všech formách života, a neexistují důkazy pro původ apoptózy evolucí. Ačkoli se systém trochu mění u různých forem života, živý tvor má buď kompletní, složitý, fungující systém apoptózy nebo ji zcela postrádá. Výzkum apoptotické molekulární mašinérie ukázal, že všechny architektury proteinové domény jsou založeny na týchž „vysoce konzervovaných“ doménách. Tento termín „vysoce konzervovaný“ prostě znamená, že kód DNA užitý při výrobě proteinu je velmi podobný jak v takzvaných „primitivních“ tak v „pokročilých“ formách života (Aravind et al., 2001) – fakt, který silně podporuje závěr, že tyto základní domény byly naplánovány. V darwinovských termínech, mechanizmus apoptózy je „evolučně konzervován“ (Shi, 2002, str. 459). To znamená, že podobný systém nalézáme ve všech formách života, jak bychom očekávali, kdyby byly všechny stvořeny.
Apoptóza také podporuje předpoklad neredukovatelné složitosti, který postuluje, že jistá úroveň složitosti musí existovat, aby systém vůbec fungoval (Elmore, 2007). Protože proces apoptózy je podobný u všech organizmů, očekávali bychom, že významná podobnost existuje mezi geny lidské apoptózy a geny apoptózy takzvaných „primitivních“ či „jednoduchých“ zvířat. Očekávali bychom, že bílkoviny, jež mají podobné funkce, mají velmi podobné plány u všech organizmů od červů po člověka, a tak to též nalézáme. Systém apoptózy je dalším příkladem podrobné sady neredukovatelně složitých biochemických mechanizmů, které nemají darwinistické vysvětlení (Behe, 1996).
Všechny z 200 000 odhadovaných lidských bílkovin musí náležitě spolupracovat, aby každá mohla vykonávat stanovenou funkci a nebránit ve fungování dalším bílkovinám či buněčným procesům. Je-li jediná bílkovina změněna, může to mít neblahé důsledky na celou buňku. Pokud změna způsobí, že potřebná bílkovina nefunguje, funkce buňky bude omezena, a to patrně vyústí do změněné bílkoviny, která bude bránit ve fungování dalších struktur (Zubay et al., 1995; Yockey, 1992; Branden a Tooze, 1999). Toto zřetězení událostí platí obzvlášť pro systémy určené zřejmě ke zničení buňky, jako třeba apoptózu.
Objev apoptózy mluví proti neodarwinizmu také proto, že tento mechanizmus je pro buňku smrtelný, pokud celá ta spousta překážek a rovnováh není na svém místě jako kompletní sada. Enzymy a mechanizmy, které apoptóza užívá ke zničení buňky, pokud nejsou přísně kontrolovány, se mohou snadno zvrhnout a poškodit či zabít normální zdravé buňky, jak se někdy skutečně děje. Proto je obtížné předvídat darwinovský mechanizmus, jenž umožní mutacím vyvinout systém apoptózy, a přitom udržet tento systém pod řádnou kontrolou a v řádném fungování tak, aby neustále neničil právě ony populace, pro něž je apoptóza prý vyvinuta jako přínos. Podobně jako u evoluce opravných mechanizmů DNA (Bergman, 2005), aktivita apoptózy by byla v rozporu s týmž evolučním mechanizmem, který ji údajně zkonstruoval.
Apoptóza je asi jedním z důkazů proti hypotéze sobeckých genů, jež postuluje, že účelem genu je vposledku pouze vlastní sebezáchova. Podle tohoto názoru, rostlinná a zvířecí těla jsou pouze stroji k přežívání „vytvořenými našimi geny“, a „dominující kvalitou, kterou bychom očekávali od úspěšného genu, je nemilosrdné sobectví“ (Dawkins, 1976, str. 2). Tohle nemilosrdné sobectví genů znamená, že „se starají“ pouze o vlastní propagaci, a vposledku užívají celé tělo jen pro svůj vlastní cíl. Slovy Wilsona: „Organizmus je pouze cestou DNA k tomu, jak nadělat více DNA“ (1975, str. 3).
Více než kterýkoli jiný současný vědec zpopularizoval Dawkins myšlenku, že všechny živé věci jsou pouhými dopravními prostředky pro geny, jejichž jediným biologickým účelem je propagace vlastní DNA. Jeho základní tezí je, že geny „se rojí v obrovských koloniích…bezpečné uvnitř gigantických vlekoucích se robotů, odizolovány od vnějšího světa, a přece jím manipulující pomocí dálkové kontroly. Jsou ve vás i ve mně; vytvořily naše tělo i mysl; a jejich zachování je zásadním důvodem pro naši existenci…my (fenotypy čili individuální organizmy) jsme stroji, které slouží jejich přežívání“ (Dawkins, jak citován v Milnerovi, 1990, str. 402).
Tato teze o genovém selekcionizmu je tak široce přijímána, že „je často označována prostě jako moderní darwinizmus“ (Johnson, 2000, str. 106). Dawkins učí, že všechny aktivity života vposledku existují za účelem zachování a rozmnožení nemilosrdně sobeckých genů.
Evoluce je velmi pružnou teorií. Ačkoli darwinisté uznávají, že neodarwinizmus by normálně měl sklon upřednostňovat přežití každé buňky, aby mohla předat své geny , někteří z nich se pokusili vysvětlit apoptotický mechanizmus v rámci hypotézy sobeckých genů (Dawkins, 1976). Dawkins poznamenal, že zemřou-li nemocné buňky, organizmus jako celek má skvělou výhodu delšího života, a tím předání svých genů potomstvu. Jedním z problémů tohoto vysvětlení původu apoptózy je, že apoptóza není činná v mnoha buňkách dříve než uplyne reprodukční věk organizmu. Jiným problémem je, že mnoho z četných buněk zničených apoptózou neovlivňuje schopnost organizmu přežít. Jiné metody, které by byly v harmonii s teorií sobeckých genů, by mohly dosáhnout téhož cíle, třeba přítomnost propracovanějšího opravného systému a využívání větší ochrany proti poškození buňky, takže méně buněk by muselo být zničeno.
Tělo produkuje mnoho struktur odstraňováním buněk, což činí tělo, nikoli DNA, biologicky nejdůležitějším. Pokud by byla DNA biologicky důležitější, byl by býval využit spíše prostředek k vyvinutí požadovaného rysu embryologicky – prostředek, který by spíše ladil s teorií sobeckých genů, než aby se spoléhalo na apoptózu, jež by odstranila jisté buňky poté, co se vyvinuly. Apoptóza je vlastně špatnou volbou, která musí být překlenována, aby mohla buňka pokračovat ve své mitotické dráze. Pokud není tato cesta přepsána příslušným signálem, buňka vždy nastoupí apoptózu. To je v rozporu s tím, co bychom očekávali od neodarwinistického původu systému.
Mezi mnoha dalšími problémy, jimž čelí Dawkinsova teorie sobeckých genů, je fakt, že geny samy o sobě jsou bezcenné. Je to pouze software, a bez buněčné mašinérie, které se účastní stovky enzymů a složitých struktur jako třeba ribozomy („hardware“), by život nemohl fungovat. Geny jsou jenom částí celku umožňujícího přežití, a pro toto přežití je třeba složitá buněčná organizace jako celek (Morange, 2002). Staré ústřední dogma bylo: DNA – RNA – bílkoviny. Nové dogma je: genom jako celek – genové produkty – struktura a funkce – dráhy a fyziologie (Morange, 2002), což se vše hodí do koncepce původu stvořením.
Geny jsou též vypínány a kontrolovány negenetickými faktory – procesem zvaným epigenetická kontrola. Kompenzace množství chromozomu X u žen, kde je jeden ze dvou X chromozomů v každé somatické buňce náhodně vypnut metylací v raném stadiu vývoje, je jedním z těchto příkladů. Margulis a Sagan (2002) rezolutně vystupují proti hypotéze sobeckých genů tvrzením, že celá biosféra spolupracuje do té míry, že funguje jako celek, koncepce zvaná hypotéza Gaia (2002). Prohlašují, že gen není nikdy „osobností“, ale „pouze kouskem DNA dost dlouhým na to, aby fungoval“ a že „přišel čas v seriózní biologii k opuštění slov jako „sobecké geny“ a jejich nahrazení plnohodnotnými termíny“ (Margulis a Sagan, 2002, str. 16-17).
Současný výzkum zjistil „podstatně“ větší míru složitosti u genomu než se myslelo ještě před pouhým desetiletím (Aravind et al., 2001). Apoptóza je nesmírně složitý genetický a biochemický systém, který je pouze jedním příkladem z mnoha, kde geny dávají přednost organizmu, ne obráceně, jak předpovídá Dawkinsův model sobeckých genů. Ani zárodečná buněčná linie ani somatická buněčná linie příslušné buňky z toho přitom nemají prospěch, protože apoptóza ničí obě navždy; takže jedině organizmus z toho může mít prospěch. Často neumírají pouze podružné buňky, jak to učí neodarwinizmus, ale buňky, jež jsou buď škodlivé pro organizmus, nebo buňky, které jsou nepotřebné, hynou, protože jsou na špatném místě ve špatnou dobu (Elmore, 2007).
A dále, evoluce není schopna vysvětlit původ tohoto systému, u něhož analýza prokázala, že jak mechanizmus tak zúčastněné geny jsou velmi podobné u všech dosud studovaných eukaryont (Elmore, 2007). Tím je podpořeno tvrzení, že apoptóza je neredukovatelně složitá. Apoptóza je nesmírně složitá genetická a biochemická organizace, která je pouze jedním z mnoha tisíc příkladů neredukovatelně složitého systému, který reguluje život buňky pro zdraví organizmu jako celku.
Chtěl bych poděkovat Johnu Woodmorappovi, M. S. , Waynu Frairovi, Ph. D., Cliffordu Lillovi, M.S., Bertu Thompsonovi, Ph. D., Kevinu Andersonovi, Ph. D., Georgu Howemu, Ph. D., a několika anonymním recenzentům za jejich komentáře k prvním verzím tohoto rukopisu.
Abeles,
R.H., P.A. Frey, and W.P. Jenks.
1992. Biochemistry.
Jones and Bartlett,
Boston,
MA.
Adams,
J.M., and S.Cory. 1998. The Bcl-2
protein
family: arbiter of cell survival.
Science
281:1322–1325.
Alberts,
B. 2004. Essential Cell Biology.
Garland
Sience, New York, N.Y.
Aravind,
L., V.M. Dixit, and E.V. Koonin.
1999. The
domains of death: evolution
of the
apoptosis machinery. Trends in
Biochemical
Sciences 24:47–53.
Aravind,
L., V.M. Dixit, and E.V. Koonin.
2001.
Apoptotic molecular machinery:
vastly
increased complexity in vertebrates
revealed
by genome comparisons. Science
291:1279–1283.
Ashkenazi,
A., and V.M. Dixit. 1998. Death
receptors:
signaling and modulation.
Science
281:1305–1308.
Barinaga,
M. 1998. Death by dozens of cuts.
Science
280:32–34.
Behe,
M. 1996. Darwin’s Black Box. The
Free
Press, New York, NY.
Bergman,
J. 1998. The unbridgeable chasm
between
prokaryotes and eukaryotes,
pp.
67–79 in The Fourth International
Conference
on Creationism. Edited by
R.E.
Walsh. Pittsburgh, PA.
Bergman,
J. 2005. The mutation repair systems:
A major
problem for macroevolution.
Creation Research Society Quarterly
41:265–273.
Blackstone,
N.W., and D.R. Green. 1999.
The
evolution of a mechanism of cell
suicide. Bioassays
21:84–88.
Branden,
C., and J. Tooze. 1999. Introduction
to
Protein Structure. Garland, New
York, NY.
Dawkins,
R. 1976. The Blind Watchmaker.
Norton,
New York, NY.
Dexter,
T.M., M.C. Raff, and A. H. Wyllie
(Ed).
1995. The Role of Apoptosis in
Development,
Tissue Homeostasis and
Malignancy.
Chapman and Hall, New
York, NY.
Duke,
R.C; D.M. Ojcius, and J.D.E. Young.
1996. Cell
suicide in health and disease.
Scientifi
c American 275:80–87.
Elmore,
S. 2007. Apoptosis: a review of
programmed
cell death. Toxicologic
Pathology
35(4):495–516.
Fesik,
S.W. 2000. Insights into programmed
cell death
through structural biology.
Cell 103:273–282.
Golstein,
P., D.M. Ojcius, and J.D.E. Young.
1991. Cell
death mechanisms and the
immune
system. Immunological Reviews
121:
29–65.
Gregory,
C.D. (Editor). 1995. Apoptosis and
the Immune Response. Wiley-Liss, New
York, NY.
Gross, A., J.M. McDonnell, and S.J. Korsmeyer.
1999. BCL-2 family members
and the mitochondria in apoptosis.
Genes and Development 13:1899–1911.
Hartwell, L., and M. Kastan. 1994. Cell
cycle control and cancer. Science
266:1821–1827.
Hartwell, L., and T.A. Weinert. 1989. Checkpoints:
controls that ensure the order of
cell cycle events. Science 246:629–633.
Hengartner, M.O. 1998. Death cycle and
Swiss army knives. Nature 391:441–
450.
Hengartner, M.O. 1998a. Injected cytochrome
C induces apoptosis. Nature
391:449–450.
Hengartner, M.O. 2000. The biochemistry of
apoptosis. Nature 407:770–775.
Huang, D.D.C., and A. Strasser. 2000. BH3-
only proteins—essential initiators of
apoptotic cell death. Cell 103:839–842.
Johnson, P. 2000. The Wedge of Truth. Inter-
Varsity Press, Downers Grove, IL.
Kerr, J.F.R., A. H. Wyllie, and A.R. Currie.
1972. Apoptosis: a basic biological phenomenon
with wide-ranging implications
in tissue kinetics. British Journal of
Cancer 26: 239–257.
Korsmeyer, S.J. 1995. Regulators of cell
death. Trends in Genetics 11:101–105.
Krakauer, D.C., and R.J. Payne. 1997. The
evolution of virus-induced apoptosis. Proceedings
of the Royal Society of London.
Series B: Biological 264:1757–1762.
Kroemer, G. 1997. Mitochondrial implication
in apoptosis: towards an endosymbiont
hypothesis of apoptosis evolution Cell
Death and Differentiation 4:443–456.
Lewin, B. 1997. Genes VI. Oxford Oxford
University Press, New York, NY.
Lewis, R. 2000. Genetics. McGraw Hill, New
York, NY.
Maddika, S., S.R. Ande, S. Panigrahi, T.
Paranjothy, K. Weglarczyk, A. Zuse, M.
Eshraghi, K.D. Manda, E. Wiechec, and
M. Los. 2007. Cell survival, cell death
and cell cycle pathways are interconnected:
implications for cancer therapy.
Drug Resistance Updates 10:13–29.
Margulis, L., and D. Sagan. 2002. Acquiring
Genes; A Theory of the Origin of Species.
Basic Books, New York, NY.
Martin, S.J., and D.R. Green. 1995. Protease
activation during apoptosis: death by a
thousand cuts? Cell 82:349–352.
McColl, A., S. Michlewska, I. Dransfi eld,
and A.G. Rossi. 2007. Effects of glucocorticoids
on apoptosis and clearance
of apoptotic cells. The Scientifi c World
Journal 7:1165–1181.
Medina, J.J. 1996 The Clock of Ages. Cambridge
University Press, New York, NY.
Mercer, J., M. Mahmoudi, and M. Bennett.
2007. DNA damage, p53, apoptosis and
vascular disease. Mutation Research
621:75–86.
Milner, R. 1990. The Encyclopedia of Evolution.
Facts on File, New York, NY.
Morange, M. 2001. The Misunderstood Gene.
Harvard University Press, Cambridge,
MA.
Affara, M., B. Dunmore, C. Savoie, S. Imoto,
Y. Tamada, H. Araki, D.S. Charnock-
Jones, S. Miyano, and C. Print. 2007.
Understanding endothelial cell apoptosis:
what can the transcriptome, glycome and
proteome reveal? Philosophical Transactions
of the Royal Society of London Biology
Science. 362(1484):1469–87
Raff, M.C. 1996. Death wish. The Sciences
36(4):36–40.
Rupinder, S.K., A.K. Gurpreet, and S. Manjeet.
2007. Cell suicide and caspases.
Vascular Pharmacology 46:383–393.
Sharon, J. 1998. Basic immunology. Williams
and Williams, Baltimore, MD.
Shi, Y. 2002. Mechanisms of caspase activation
and inhibition during apoptosis.
Molecular Cell 9:459–470.
Steller, H. 1995. Apoptosis: mechanisms
and genes of cellular suicide. Science
267: 1445–1449.
Veggeberg, S.K. 1995. Cell suicide research
may yield life-saving cures. BrainWork
5:4–6.
Wang, X.W, H. Yeh, L. Schaeffer, R. Roy,
and V. Moncollin. 1995. P53 modulation
of TF11H associated nucleotide
excision repair activity. Nature Genetics
10:188–195.
Weinrauch, Y., and A. Zychlinsky. 1999.
The induction of apoptosis by bacterial
pathogens. Annual Review of Microbiology
53:155–187.
Wiens, M., A. Krasko, C.I. Muller, and W.E.
Miller. 2000. Molecular evolution of
apoptotic pathways: cloning key domains
from sponges (Bcl-2 homology domains
and death domains) and their phylogenetic
relationships. Journal of Molecular
Evolution 50:520–531.
Williams, G.T. and C.A. Smith. 1993.
Molecular regulation of apoptosis:
genetic controls on cell death. Cell
74:777–779.
Wilson, E.O. 1975. Sociobiology: the New
Synthesis. Harvard University Press,
Cambridge, MA.
Wu, J. 1996. Apoptosis and angiogenesis:
two promising tumor markers in
breast cancer. Anticancer Research 16:
2233–2239.
Yockey, H. 1992. Information Theory and
Molecular Biology. Cambridge University
Press, New York, NY.
Zamzami, N., and G. Kroemer. 2001.
The mitochondrion in apoptosis: how
pandora’s box opens. Nature Reviews
2:67–71.
Zubay, G. 1995. Principles of Biochemistry.
Wm C. Brown, Dubuque, IA.